Fig. 9.1
ECG showing atrial fibrillation with rapid ventricular response
The echocardiographic examination showed severely dilated left ventricle (LV end-diastolic volume of 280 ml, LV end-diastolic diameter 78 mm), impaired LV function with an estimated ejection fraction (EF) of 20 % because of global hypokinesia, correct function of the mitral prosthesis with a mild intraprosthetic regurgitation, right ventricle dilatation and dysfunction (TAPSE 12 mm, FAC area 25 %), and severe tricuspid regurgitation with systolic pulmonary artery pressure of 50 mmHg (Fig. 9.2). The cardiac output and the cardiac index as estimated by echocardiography were, respectively, 2.8 l/min and 1.3 l/min/m2.
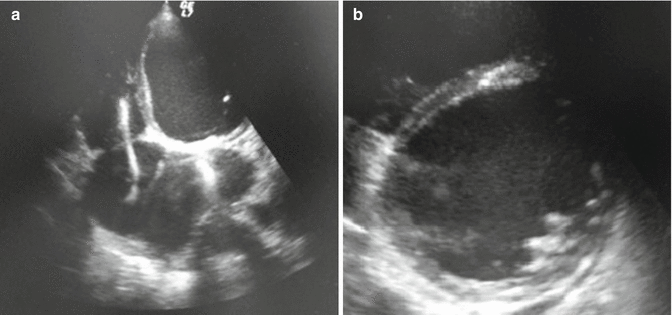
Fig. 9.2
Echocardiographic images: apical four-chamber view (a) and parasternal short axis at papillary level (b) showing chamber dilation, mitral valve prosthesis, and presence of lead in right chambers
Urgent chest radiography was performed showing pulmonary vascular congestion with alveolar infiltrates and right moderate pleural effusion.
Clinical Course and Therapeutic Management
The patient underwent insertion of a radial arterial catheter for invasive measurement of blood pressure and arterial blood gas testing, insertion of central venous catheter at right subclavian site for drug infusions, measurement of central venous pressure and blood sampling, and determination of central venous oxygen saturation.
Clinical, instrumental, and laboratory data (hypotension <90 mmHg, tissue hypoperfusion with increase in arterial lactate and renal and hepatic impairment, cardiac index of 1.3 l/min/m2) allow us to make the diagnosis of cardiogenic shock in a patient with advanced heart failure.
Inotropic support with dopamine (5 mcg/kg/min) was continued, and also adrenaline was started at a dose of 0.05 up to 0.08 mcg/kg/min because of persistent hypotension, increase in arterial lactate (7.8 mmol/l), and oliguria.
Continuous infusion of loop diuretics (furosemide, 500 mg/24 h) was started with progressive improvement in diuresis. Intravenous digoxin was administered in the acute phase for AF rate control, and unfractionated heparin was introduced. Insulin infusion was started to correct hyperglycemia and on the following days switched to bolus insulin injection.
The patient’s clinical status gradually improved, with normalization of arterial lactate on day 2 and also the central venous oxygen saturation (from baseline 56–63 % on day 2). On day 5, pulmonary congestion significantly improved and the patient was extubated. The patient was gradually weaned from inotropes until interruption on day 7. Therefore, an echocardiography was repeated confirming severe left ventricular dysfunction (FEVS 25 %) and mild to moderate right ventricle dysfunction (TAPSE 15 mm, FAC area 32 %), reducing pulmonary artery pressure (35 mmHg).
A beta-blocker and an angiotensin-converting enzyme inhibitor were started on day 8 and up-titrated (ramipril 2.5 mg o.d., bisoprolol 3.75 mg o.d.). Loop diuretics were switched from IV to oral administration (furosemide 125 mg b.i.d., spironolactone 100 mg o.d.). Oral digoxin was continued to achieve a better rate control. Oral anticoagulation was reintroduced on day 9. Laboratory tests showed improvement of kidney and hepatic function (creatinine 1.4 mg/dl, total bilirubin 1.3 mg/dl, normalization of AST, ALT, and γGT).
Cardiac rehabilitation was started on day 12 with progressive improvement of functional capacity.
Considering the end-stage HF despite optimal pharmacological and device treatment and the recent episode of cardiogenic shock, the possibility to an evaluation for advanced treatment options, as heart transplant or left ventricular assistive device implantation, was offered to the patient who agreed, and a visit in a national reference hospital was therefore planned. The patient was discharged on day 25.
9.2 Cardiogenic Shock
Definition and Epidemiology
Cardiogenic shock (CS) is a complex clinical condition characterized by inadequate end-organ perfusion due to the inability of the heart to provide adequate flow. The tissue hypoperfusion, if prolonged, could result in end-organ damage and finally in multiorgan failure. Cardiogenic shock is a fatal condition if not early diagnosed and treated. The in-hospital mortality approaches 50 % and is related to the severity of hemodynamic impairment, the promptness of diagnosis, and the type of management (medical therapy, mechanical support) [1]. Mortality decreased significantly during the last years because of the wide use of revascularization.
The diagnosis of CS results from multiparametric evaluation and could be made in the presence of:
Hypotension defined as systolic blood pressure ≤90 mmHg or when vasopressors are required to maintain SBP ≥90 mmHg or mean arterial pressure is 30 mmHg lower than baseline
Evidence of organ hypoperfusion: resting tachycardia, altered mental status, oliguria, poor capillary refill, cold/diaphoretic extremities
A reduction in cardiac index (<1.8 L/min/mq without support or <2.2 l/min/mq with support) with evidence of increase in pulmonary capillary wedge pressure (>18 mmHg)
Any cause of severe left or right ventricle dysfunction may cause cardiogenic shock; however, acute coronary syndrome (ACS) with left ventricular failure is mostly involved. The incidence of CS complicating ACS is approximately 7 % in ST elevation myocardial infarction (STEMI) and 2.5 % in non-STEMI [2]. In those patients presenting with ACS and CS, mechanical complications as ventricular septal or free wall rupture and papillary muscle rupture should be suspected and searched.
Other less frequent causes are acute myopericarditis, stress-induced cardiomyopathy, acute valvular regurgitation or prior severe valvular disease, hypertrophic cardiomyopathy, dilated cardiomyopathy, drugs and medications, arrhythmias, and traumatic cardiac injury.
Cardiogenic shock due exclusively to right ventricle involvement represents only 5 % of cases, and it is characterized by a similar mortality to LV shock [3].
In the ischemic setting, CS could be present acutely or could develop later, within the first days. It seems that later CS is associated with a higher mortality than earlier development of CS [4].
Different risk factors for CS in ischemic patients have been recognized: anterior STEMI, multivessel disease, advanced age, female sex, previous diagnosis of diabetes and hypertension, prior cardiovascular disease, heart failure at admission, systolic blood pressure <120 mmHg, heart rate >90 bpm, and presence of left branch block [5, 6].
Pathophysiology
Systemic perfusion and blood pressure are related to cardiac output (CO) and systemic vascular resistance (SVR):
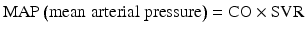
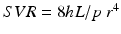
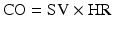
η = viscosity, L = vessel length, r = vessel radius, SV = stroke volume, HR = heart rate
The stroke volume depends on the preload, afterload, and myocardial contractility as explained by the Frank–Starling and Hill mechanisms. The initial response to a decrease in blood pressure is mediated by arterial baroreceptors that cause an enhancement in sympathetic activity (via IX and X cranial nerves) with a consequent increase in HR, myocardial contractility, and SVR. More slowly acting mechanisms are the activation of renin/angiotensin/aldosterone system and fluid retention. The reduction in tissue perfusion leads to a reduced oxygen delivery with a shift to anaerobic metabolism and an increase in lactate levels with a possible consequent metabolic acidosis.
Cardiogenic shock (CS) may be precipitated by different cardiac and extracardiac causes as listed below:
Cardiomyopathies: acute myocardial infarction (MI) involving >40 % of the left ventricular myocardium or dilated cardiomyopathy with cardiac pump failure.
Arrhythmias: supraventricular arrhythmias may cause cardiogenic shock through an impairment of left ventricular filling. Bradyarrhythmias or ventricular tachycardia/fibrillation may reduce or abolish CO due to an ineffective cardiac contraction.
Mechanical: valvulopathies (mitral or aortic regurgitation) or intracardiac shunt.
Extracardiac: any condition that causes a significant reduction in preload or acute increase in afterload (i.e., cardiac tamponade, pulmonary embolism, tension pneumothorax, constrictive pericarditis).
The CS pathophysiology is very complex with differences from patient to patient. Cardiogenic shock evolves through different stages that represent a physiologic continuum from an initially compensated status (pre-shock or shock impending) till multiorgan failure.
Regardless of the precipitating cause, the main feature consists of a reduction of cardiac output with consequent hypotension, unable to maintain an adequate systemic perfusion (Fig. 9.3). The reduction of blood pressure triggers the activation of compensatory mechanisms through sympathetic system and renin/angiotensin/aldosterone system with consequent tachycardia, increased contractility, a marked systemic vascular resistance (SVR) elevation increasing LV afterload, and fluid retention with increase in preload. These compensatory mechanisms in the long term become maladaptive and result in a further marked reduction in tissue perfusion. Hypotension, vasoconstriction, tachycardia, and increased myocardial contractility reduce myocardial perfusion and increase myocardial oxygen demand, exacerbating ischemia.
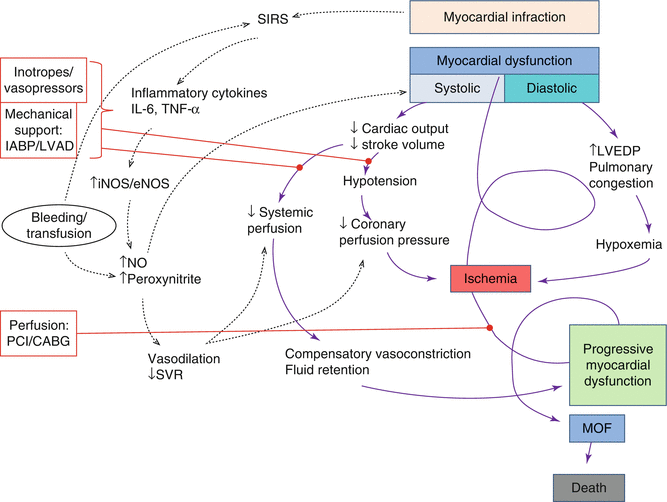
Fig. 9.3
Concept of CS pathophysiology. This is the “downward spiral,” induced by left ventricle (LV) systolic dysfunction that leads to reduced stroke volume and cardiac output with consequent hypotension. Coronary blood flow is therefore reduced with ischemia and further myocardial dysfunction. Even diastolic dysfunction with increased left ventricular end-diastolic pressure (LVEDP) and pulmonary edema leads to hypoxemia and consequent ischemia. The reduced systemic perfusion activates compensatory mechanisms that cause vasoconstriction and fluid retention, increasing left ventricle after- and preload and aggravating myocardial dysfunction. The systemic inflammatory response syndrome (SIRS) characterized by cytokine (interleukin-6 (IL-6) and tumor necrosis factor (TNF)) production and consequent endothelial and inducible nitric oxide synthase (eNOS, iNOS) activation leads to nitric oxide (NO) and peroxynitrite production that causes reduced systemic vascular resistance (SVR) with vasodilatation and further myocardial depression. Bleeding complications and subsequent transfusions have a negative role in the shock spiral. If there is not a prompt intervention with treatment options shown in red (inotropes/vasopressors, mechanical support with intra-aortic balloon pumping (IABP) and left ventricular assist device (L-VAD), reperfusion by percutaneous coronary intervention (PCI) or coronary artery bypass (CABG)), the vicious circle leads to multiorgan failure (MOF) and death
Pump failure causes diastolic dysfunction with increased ventricular diastolic pressure that further reduces coronary perfusion pressure, worsening ischemia. The increased ventricular diastolic pressure increases left atrial pressure which may cause pulmonary congestion leading to hypoxia, further exacerbating myocardial ischemia. Ischemia worsening aggravates myocardial dysfunction and begins a vicious cycle that leads to progressive end-organ hypoperfusion with multiorgan failure (MOF) and death when not interrupted (Fig. 9.3). Reduced systemic perfusion leads to anaerobic metabolism and consequently to lactic acidosis that further depresses myocardial function.
Some patients do not have elevated SVR, suggesting that the compensatory vasoconstriction is not universal, and moreover, a systemic inflammatory response may be involved [7], contributing to myocardial dysfunction and hypotension (via vasodilatation). In fact, in the presence of myocardial infarct, cytokines (interleukin-6 (IL-6), tumor necrosis factor (TNF)) that activate inducible nitric oxide synthase (iNOS) are released, leading to increased levels of nitric oxide (NO) with consequent vasodilatation and worsening hypotension [8]. Nitric oxide and superoxide lead to peroxynitrite production that impairs myocardial contractility [9]. The inflammatory mediators lead to microcirculatory abnormalities like regional heterogeneity in blood flow which plays a very important role in organ failure pathogenesis. In fact data shows that IL-6 levels correlate with organ failure and mortality [10].
Even if severe LV failure is the principal cause of CS, other factors, here below listed, may contribute to hypotension:
Hypovolemia due to bleeding with a hemorrhagic shock superimposition or due to diuretic therapy
Septic shock superimposition
Severe preexistent valvular heart disease like critical aortic stenosis or new-onset valvular disease like severe mitral regurgitation
Important bradycardia that causes low cardiac output and hypotension in patients with reduced LV function due to acute MI
Atrial arrhythmias with rapid ventricular response or ventricular tachycardia
Drugs lowering blood pressure (nitrates, beta-blocker, calcium antagonists, ACE inhibitors, diuretics, and morphine)
These factors should be promptly detected and, when possible, corrected.
Diagnosis
The diagnostic evaluation during cardiogenic shock must not delay resuscitation if needed and must be conducted at the same time. Diagnostic efforts should be made to recognize the stage of pre-shock to prevent loss of the compensatory mechanisms and progression to shock and multiorgan dysfunction.
Medical History and Physical Examination
Medical history may be collected from the patient or relatives in case of advanced shock. The presence of cardiovascular risk factors or the history of chest pain may suggest acute myocardial infarction (MI). Additional information about comorbidities or allergies should be recorded.
Cardinal findings on physical examination are:
Hypotension: defined as absolute (PAS <90 mmHg or PAM <65 mmHg) or relative (ΔP > 30 mmHg). Prominent and persistent hypotension (>30 min), despite volemic correction, may require inotropes to ensure adequate systemic perfusion.
Oliguria: decreased urine output (diuresis <0.5 mg/kg/h), consequence of renal hypoperfusion related to reduced cardiac output and blood redistribution to other vital organs.
Cool and clammy skin: compensatory vasoconstriction to redirect blood flow to vital organs, causes cold, mottled, or diaphoretic skin.
Altered mental status: ranges from agitation to delirium and coma.
Dyspnea, chest pain with tachycardia, and tachypnea are often present. On cardiac auscultation, gallop rhythm or new murmurs may be found. Pulmonary congestion with diffuse crackles is also a typical finding but may lack in about one-third of patients at presentation [11]. Jugular venous distension and hepatomegaly are clinical signs related to an increased preload, especially during right ventricular failure. A capillary refill time >2 s is a frequent finding and should be associated with low mixed venous oxygen saturation.
Electrocardiogram
Electrocardiogram (ECG) suggests the diagnosis of acute MI in the presence of ST-T alterations. Supraventricular and ventricular tachy- or bradyarrhythmias may cause shock and can be diagnosed by ECG monitoring. Shocked patients usually present sinus tachycardia.
Echocardiogram
Echocardiography may confirm the diagnosis of cardiogenic shock, showing marked depression of left or right ventricular function with low stroke volume and elevated filling pressures. It is also useful in evaluating cardiac chambers, regional wall motion, the pericardium, and valves: it could detect causes or contributing factors as regional wall motion abnormalities, the presence of cardiac tamponade, or severe mitral or aortic regurgitation. In acute MI, echocardiogram should be repeated to exclude the presence of mechanical complications as ventricular septal, free wall, or papillary muscle rupture [12]. Transthoracic echocardiography (TTE) is the first step, but transesophageal echocardiography (TEE) should be used when TTE images are suboptimal especially in patients with mechanical ventilation.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


