Fig. 12.1
Correlated gene groups. (a) Cluster dendrogram showing 13 correlated gene groups. Clustering was derived by cutting the cluster tree at the 0.001 quantile of a random distribution. The cluster distances are indicated on the y-axis. (b, c) Example of two correlated gene groups showing highly correlated patterns of expression in samples of patients and healthy individuals. Centered expression vectors were sorted by defined meta-phenotypes (Figure adapted from Toenjes et al. [5])
In a study by Schlesinger et al., the global transcription network driven by Gata4, Mef2a, Nkx2-5, and Srf (serum response factor) was investigated in the mouse cardiomyocyte cell line HL-1. ChIP-chip data demonstrated the combinatorial regulation by these TFs and RNAi knockdown experiments showed that they partially compensate each other’s function. In detail, genes bound by multiple TFs in their promoter and/or enhancer region are less likely differentially expressed in the knockdown of one respective factor, demonstrating the buffering capacity of the network [6]. Finally, the genome-wide combinatorial binding of Gata4, Nkx2-5, Tbx5, Srf, Mef2a, as well as the histone acetyltransferase p300 determined by ChIP followed by next-generation sequencing (ChIP-seq) in HL-1 cells has also been used to identify cardiac enhancer regions. Interestingly, the majority of loci bound by multiple cardiac TFs did not overlap with loci bound by p300 [7], a transcriptional coactivator commonly used to identify enhancers [8].
To dissect the spatiotemporal protein networks driving human heart development, a recent study constructed functional clusters from over 250 cardiac development genes based on morphological subgroups corresponding to the specific phenotype associated with their mutation [9]. In general, increased complexity of the heart during development is accompanied by a higher complexity at the molecular level. This is reflected by a higher number of functional modules in networks associated with “late phenotypes” compared to “early phenotypes.” The TFs GATA4, TBX5, and NKX2‐5 were found to be involved in the majority of networks in almost all stages of cardiogenesis, demonstrating their central role in the cardiac transcription network. Interestingly, the respective modules show a high degree of variation in their complexity and protein composition, leading to a flexible and combinatorial regulation by the core cardiac transcription factors. Taken together, these results underline the dynamics of cardiac transcription networks and are consistent with the striking phenotypic variability that can result from mutations of a single factor [10, 11].
12.2 Core Myogenic Transcription Factors
12.2.1 Nkx2-5
The transcription factor Nkx2-5 (NK2 homeobox 5) is essential for multiple aspects of heart development [12]. It is expressed in early heart progenitor cells and expression remains high in adult hearts [13]. Nkx2-5 physically interacts with other core cardiac TFs in a combinatorial manner. For example, Gata4, Tbx5, and Nkx2-5 synergistically activate the ANF promoter [14]. Moreover, Nkx2-5 interacts with Hand2 and Mef2c to determine ventricular identity [15, 16]. In combination with Srf and Gata4, it further activates the expression of sarcomeric genes [17]. Mice lacking Nkx2-5 show abnormal morphogenesis of the heart tube as well as failure of left ventricular development and die early during embryonic development (lethality at E9.5) [18]. In addition, Nkx2-5 functions in the development and maintenance of the conduction system, demonstrated by progressive atrioventricular (AV) block observed in Nkx2-5 heterozygous mice [19]. NKX2-5 mutations in humans account for approximately 4 % of all CHD cases including AV block, septal defects, TOF, left ventricular noncompaction, double outlet right ventricle (DORV), AS, and Ebstein’s anomaly [20, 21].
12.2.2 Mef2
Myocyte enhancer factor 2 (Mef2) proteins are a family of TFs that play a central role in cellular differentiation. They contain both a highly conserved MADS box and an immediately adjacent motif, the MEF2 domain, which together mediate dimerization, DNA binding, and cofactor interactions [22]. The MEF2 genes MEF2A, MEF2B, MEF2C, and MEF2D are all expressed in every stage of the developing human heart [23] and are crucial for the expression of muscle-specific genes [24]. They interact with other key cardiac TFs, particularly to regulate the expression of contractile proteins via interacting with members of the MyoD (myogenic differentiation) family, which play a key role in regulating muscle differentiation. For example, Mef2c regulates expression of Dpf3 (D4, zinc and double PHD fingers, family 3, also known as BAF45c) [25], a heart- and muscle-specific subunit of the BAF chromatin remodeling complex, and moreover interacts with Tbx5 to activate the expression of the Myh6 (cardiac alpha-myosin heavy chain 6) gene [26]. Within cardiac muscle lineages, the expression of Mef2 itself is in turn regulated by NK2 proteins [27]. Moreover, the different Mef2 proteins can partly compensate each other’s functions [28]. In addition to other TFs, Mef2 proteins can also interact with HATs (histone acetyltransferase, e.g., p300) and HDACs (histone deacetylase, e.g., HDAC4) to activate or repress the transcription of downstream targets [29]. HDAC4 expression is in turn repressed by the myogenic microRNA miR-1, which leads to an activation of Mef2 that together with MyoD initiates miR-1 expression in a feedback loop [30].
Mef2a is predominantly expressed in postnatal cardiac muscle cells, and mice lacking Mef2a die within the first week after birth due to myofibrillar fragmentation, mitochondrial disorganization, and impaired myocyte differentiation [31]. Mutations in Mef2a identified in patients implicate its signaling pathway in the pathogenesis of coronary artery disease and myocardial infarction [32]. In Mef2c null mice, failure to undergo cardiac looping and formation of the right ventricle have been reported [33], and a Mef2c enhancer has been defined that drives gene expression in the second heart field and has been widely used for conditional gene inactivation [34].
12.2.3 Tbx
Tbx proteins are characterized by a conserved 180 amino acid long region termed the T-box [35]. Members of this family including Tbx1, Tbx2, Tbx3, Tbx5, Tbx18, and Tbx20 are involved in early cardiac lineage determination, chamber specification, and the development of the conduction system [36]. Tbx2 and Tbx3 act redundantly during outflow tract (OFT) development and repress Tbx1 expression in ventral pharyngeal endoderm, while their own spatial expression is regulated by a feedback loop by Tbx1. Thus, the three factors form a crucial regulatory network for pharyngeal and OFT development, which is demonstrated by severe cardiac defects caused by knockout of Tbx1 and either Tbx2 or Tbx3 [37]. Tbx5 is involved in multiple pathways and combinatorial interactions with other TFs [38]. For example, it physically interacts with Nkx2-5 to control gene expression in cells of the cardiac conduction system [39] or forms a transcriptional complex with Mef2c required for early heart development [26]. Finally, Tbx20 interacts with Gata4 to activate both Mef2c and Nkx2-5 enhancers [40].
Heterozygous Tbx5 knockout mice suffer from septation defects and AV block, similar to heterozygous Nkx2-5 knockout mice [39]. Similar to the human GATA4 mutant phenotype, double heterozygous Tbx5 and Gata4 knockout mice show atrioventricular septal defects (AVSD) and myocardial thinning [41]. In humans, TBX5 missense mutations cause Holt-Oram syndrome [42], which is characterized by heart and limb malformations. Moreover, a homozygous variation in the TBX5 enhancer abrogating the cardiac expression of the gene was observed in a patient with isolated VSD [43]. Hemizygosity of TBX1 is the major cause for the CHD phenotype in 22q11.2 deletion syndrome patients, which is characterized by conotruncal defects like TOF, DORV, and truncus arteriosus (TA) [44]. Mutations in TBX3 [45] and TBX20 [46] as well as in the TBX18 promoter region [47] have also been identified in different CHD types.
12.2.4 Gata4
The zinc finger transcription factor Gata4 (GATA binding protein 4) is essential for cardiac development and plays a critical role for embryo survival [48, 49]. The Gata family of zinc finger TFs consists of six members, which all bind the DNA sequence “A/G GATA A/T.” Gata4, Gata5, and Gata6 are expressed in the developing heart, and Gata4 and Gata6 remain expressed in adult cardiac myocytes. Both Gata4 and Gata6 regulate the expression of several cardiac-specific genes, and Gata4 is crucial for cardiac morphogenesis during mouse embryonic development [49]. Gata4 can physically interact with other cardiac TFs like Hand2 [50], Nkx2-5 [51], Mef2 [52], Srf [53], and Tbx5 [54]. In combination with Isl1 and Tbx20, Gata4 actives Mef2c and Nkx2-5 expression in the second heart field (SHF) [40]. Furthermore, Gata4 is known to regulate the transcription of several muscle-specific genes encoding contractile elements like the α- and β-myosin heavy chain (α- and β-MHC) proteins and α-actin as well as signaling molecules like atrial natriuretic factor (ANF) and brain natriuretic peptide (BNP) [55]. Gata4 DNA-binding affinity is enhanced by the HAT p300 [56], while it is negatively regulated by HDAC4, a transcriptional repressor of muscle gene expression [57]. In addition, Gata4 binds and participates in establishing active chromatin regions by stimulating histone 3 lysine 24 acetylation (H3K24ac) deposition [58].
Homozygous Gata4 null mice die between embryonic day E7.0 and E9.5 because of severe morphogenic defects in heart tube formation [59]. Cardiogenesis is sensitive to Gata4 dosage, and graded reduction of the TF results in DORV, common atrioventricular canal (CAVC), and hypoplastic ventricular myocardium [60]. In human, GATA4 mutations cause CHD including septal defects, tetralogy of Fallot (TOF), and pulmonary stenosis (PS) [12]. One GATA4 mutation described in a family with septation defects specifically disrupted the interaction with TBX5, while the binding to NKX2-5 remained unaffected, suggesting a concerted role for GATA4 and TBX5 in cardiac septation [54].
12.2.5 Hand
Hand1 and Hand2 (heart and neural crest derivatives expressed 1 and 2) are basic helix-loop-helix transcription factors, which are expressed early in derivatives of the first and second heart field (FHF and SHF), respectively [1]. They are asymmetrically expressed in the developing ventricular chambers and play an important role in cardiac morphogenesis. Hand2 knockout mice show abnormalities in the formation of the right ventricle, most probably due to loss of the SHF [61, 62]. Embryonic stem cells (ESCs) lacking Hand1 are unable to contribute to the outer curvature of the heart that gives rise to the left ventricle [63]. In developing mouse hearts, the deletion of Hand1 has been associated with CHD including ventricular septal defect (VSD), atrioventricular valve malformation, hypoplastic ventricles, and outflow tract abnormalities 1 [64]. Nkx2-5 regulates the expression of Hand1 in the FHF, and double knockout of Nkx2-5 and Hand2 ablates both ventricular chambers, leaving only an atrial remnant [1]. In human, mutations in HAND1 cause septation defects, while HAND2 mutations have been identified in patients with TOF, DORV, and PS [12].
12.2.6 Isl1
The LIM homeodomain transcription factor Isl1 (Isl LIM homeobox 1) is required for the contribution of the SHF progenitor cells to cardiogenesis [2] and directly activates the expression of Mef2c [65, 66]. Isl1 represents a key factor of the transcriptional network regulating SHF development. The central role of Isl1 for cardiac development is demonstrated by knockout mice, which completely lack the derivatives of the SHF, namely, the outflow tract, right ventricle, and much of the atria [2]. Several ISL1 SNPs have been associated with CHD in a large patient cohort [67]; however, their functional impact awaits further exploration.
The widely expressed serum response factor (Srf) is like Mef2, a MADS-box TF important for heart and muscle development. Srf is well-known to bind to a specific DNA motif, the CArG box, with the consensus sequence “CC(A/T)6GG” in promoters of its target genes [68]. Several studies demonstrated the crucial role of Srf for heart and muscle development. For example, cardiac-specific deletion of Srf in mice results in embryonically lethal cardiac defects including abnormally thin myocardium, dilated cardiac chambers, poor trabeculation, and a disorganized interventricular septum [69]. Srf autoregulates its own expression [70] and is suggested to be a master regulator of the actin cytoskeleton and contractile apparatus [68]. Since Srf is ubiquitously expressed and has relatively low intrinsic transcriptional activity, its ability to regulate cardiac transcription is highly dependent on its interaction with both positive and negative co-regulators. For example, the combinatorial expression of Gata4, Nkx2-5, and Srf directs early cardiac gene activity [17]. Moreover, Srf enhances muscle gene expression in association with myocardin (Myocd), a smooth and cardiac muscle-specific transcriptional coactivator [71]. The Srf/Myocd complex recruits p300 to Srf-binding sites, which induces histone 3 acetylation (H3ac) and actives gene expression [72]. Furthermore, a strong correlation was found between the presence of H3ac, histone 2 lysine 4 dimethylation (H3K4me2), and Srf and p300 in vivo [73]. In addition, the homeodomain protein Hopx, which does not directly bind DNA, can inhibit SRF-dependent transcription by recruiting HDAC2 to Srf target genes, thereby modulating growth and proliferation of cardiomyocytes [74]. Similar to Mef2, Srf acts in feedback loop with a microRNA (miR-133) to modulate muscle proliferation and differentiation [30]. Furthermore, Srf regulates the transcription of other microRNAs like the smooth muscle-relevant miR-143 and miR-145, which in turn cooperatively targeted a network of transcription factors including Myocd to promote differentiation and repress proliferation of smooth muscle cells [75]. In addition, Tbx1 promotes posttranscriptional regulation of Srf levels by proteasome-mediated degradation [76].
12.3 Cis-Regulatory Elements and Chromatin Structures
In humans, the number of protein-coding genes is only about 21,000, whereas the human body consists of about 1014 cells and approximately 200 different cell types, which originated from one pluripotent cell via numerous intermediate cellular stages. Thus, it seems reasonable that a large fraction of the sequence space is occupied by regulatory sequence elements defining spatiotemporal gene expression and protein functionality by “coding” for cis-regulatory elements and non-coding RNAs modulating transcription, translation, and posttranslational modification. In fact, 99 % of the human genome does not code for proteins and has been investigated by major international efforts led by the ENCODE consortium [77, 78]. Thus, a vertebrate gene is typically associated with a proximal promoter and multiple transcriptional regulatory elements (enhancers and silencers). These cis-regulatory elements are often distant from the promoter and sometimes act across intermediate genes. Forty-thousand human heart enhancers have been postulated [79], which are potential targets for DNA-binding transcription factors and consist of sequence motifs recognized by them. The specific sequence of a motif and interacting proteins, RNA cofactors, local physical properties of the DNA fiber, and surrounding chromatin determine the binding affinity of TFs and thereby modulate TF function. Chromatin structure is modulated, for example, by posttranslational modifications and variants of histones, presence of DNA methylation or chromatin related proteins, and RNAs (see Chaps. 13, 14, and 15). Thus, a complex molecular network of trans– and cis-acting elements directs spatiotemporal gene expression.
12.4 MicroRNAs as Downstream Targets
Downstream targets mediate a considerable multiplication effect of transcription factor function. Only a small proportion of differentially expressed genes in loss-of-function experiments are direct targets of the respective transcription factors [6]. The transcription factor Srf represents a well-studied example for regulating muscle-specific microRNAs like the cardiac-specific miR-1 and miR-133 as well as miR-143 and miR-145 [30]. RNAi-mediated knockdown of Srf supported the role of Srf as a microRNA activator such that the majority of the differentially expressed microRNAs were found to be downregulated. The differential expression of these microRNAs explained more than two thirds of the differentially expressed downstream target genes in Srf knockdown [6]. Figure 12.2 shows the Srf-based transcription network integrating Srf-binding events, H3ac, microRNAs, and differential expression in Srf knockdown. A further and more global study investigated the relationship between tissue-specific microRNAs and TFs across multiple tissues, including the heart and skeletal muscle. Using ChIP-seq data for transcription factor binding sites (TFBS) from the ENCODE project, 2,347 interactions between TFs and tissue-specific microRNAs were identified, with the majority of the microRNAs being regulated by non-tissue-specific TFs. Moreover, TF-microRNA regulatory networks integrating verified or predicted interactions and expression data were constructed for each tissue. In the heart, the network comprises 33 TFs and 10 tissue-specific microRNAs, while in skeletal muscle 9 microRNAs are regulated by 52 TFs [80].
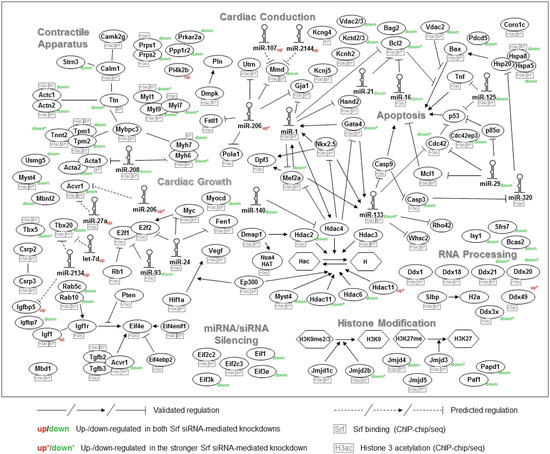
Fig. 12.2
Srf-based transcription network integrating Srf-binding events, histone 3 acetylation, microRNAs, and differential expression in Srf knockdown. The network is based on a literature search and findings from the Srf and histone 3 acetylation (H3ac), chromatin immunoprecipitation (ChIP), and Srf siRNA-mediated knockdown experiments in mouse cardiomyocytes. Srf binding and H3ac occurrence are depicted in small boxes and up- (red) or downregulation (green) in Srf knockdown is further indicated (Figure adapted from Schlesinger et al. [6])
12.5 Disturbance of the Regulatory Network Underlying Congenital Heart Disease
More than 10 years ago, Kaynak et al. provided the first evidence that distinct types of congenital heart diseases are characterized by distinct gene expression profiles at a global scale [81]. With the development of the next-generation sequencing technology, it became feasible to study the entire exome and even the entire genome in patients. It is now apparent that the majority of CHDs, which are sporadic and nonsyndromic, are based on multiple genomic alterations, which in concert alter the molecular network [82, 83]. These are not limited to cardiac-specific factors nor to the coding part of the genome. It is highly likely that network alterations driven by alterations in non-coding sequences will emerge and have a significant impact in driving the disease state. In general, human diseases including CHDs are often caused by heterozygous genetic variants that quantitatively affect dosage of the functional encoded gene product, as it is the case for copy number variations or haploinsufficiency [84, 85]. One example is Notch1 haploinsufficiency causing bicuspid aortic valve and severe aortic calcifications in related adults. Heterozygous nonsense mutations in Notch1 cause disruption of the epigenetic architecture, which results in derepression of latent pro-osteogenic and pro-inflammatory gene networks and triggers aortic valve calcification depending on hemodynamic shear stress [85].
Insights of the impact of non-coding variations have, for example, been gained by genome-wide association studies (GWAS) assessing the association between genetic variants and a phenotypic trait of interest in a large number of individuals. The TBX3-TBX5 locus and the NKX2-5 locus frequently are implicated regions in GWAS analysis of conduction anomalies. A meta-analysis of 14 GWAS on QRS duration in individuals of European descent implicated an intronic region of the SCN10A (sodium channel, voltage gated, type X, alpha subunit) gene as a major risk region for prolonged QRS duration [86]. Intriguingly, SCN10A is located next to SCN5A, which encodes the alpha subunit of the cardiac voltage gated Na+ channel and is known to cause several types of heritable arrhythmogenic disorders. In a close examination of this risk region in SCN10A-containing rs6801957, the sentinel SNP frequently found in ECG GWAS, reveals that it is occupied by TBX3, TBX5, NKX2-5, and several enhancer-associated coactivators, such as p300 and PolII, in the adult mouse heart. Analysis of the risk allele showed that the SNP alters the sequence of the cis-regulatory motif for TBX3/TBX5, which inhibits the response of the enhancer to these factors and decreases the activity of the enhancer in zebra fish in vivo, thus potentially reducing the expression level and dosage of SCN5A [87].
Conclusion
Cardiac development is tightly controlled by transcription factors that lead to correct temporal and spatial gene expression. The core cardiac transcription factors regulate each other’s expression and can act in a combinatorial manner, resulting in a buffering capacity of the network. Moreover, co-regulators, epigenetic marks, and posttranscriptional regulators like microRNAs control their expression and functional activity. While transcription factors are clearly a main driving force for gene regulation, a number of studies now show the importance of multiple regulatory levels with a high degree of interdependency leading to a fine-tuned balance of gene expression. Disruption of cardiac transcription networks can lead to dramatic consequences for cardiogenesis and results in congenital heart disease. However, the realization of the complexity of the network and most importantly its buffering capacity at different levels provide hope that we might be able to find ways to modulate it in order to arrive at a more favorable state without the need for genetic corrections. One focus could be the role of targetable epigenetic mechanisms driven by genetic alterations or environmental factors (see Chap. 16) to reduce the burden of CHDs and most importantly improve the long-term outcome.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


