Fig. 10.1
The schematic illustrates the main structural components of a striated muscle cell sarcomere. Thin filaments (I bands) are composed of actin monomers and tropomyosin. The thick filaments (A bands) are composed of two myosin chains complexed with myosin light chains. The striated microscopic appearance of the sarcomere is due to alternating A bands and I bands. The Z lines, forming the lateral boundaries of the sarcomere, are comprised primarily of α-actinin and other proteins. The M-band in the middle of the sarcomere is formed of cross-connecting elements of the cytoskeleton. Myosin filaments are cross-linked by myomesin (This figure which originally appeared in Marian and Roberts [8] is used here with permission)
10.4 Biology of the Cardiac Troponins
The cTns are a complex of three protein subunits, troponin C (TnC, the calcium-binding subunit), troponin I (TnI, the inhibitory subunit), and troponin T (TnT, the tropomyosin-binding subunit), arranged in a 1:1:1 stoichiometric ratio. This complex plays a role in the physiologic modulation of calcium sensitivity and cardiac myocyte contraction.
As structural components of the cardiac myocyte, cTns are well conserved across species, making them a “translational biomarker” in both veterinary and human medicine and a useful indicator of drug-related cardiac damage in biomedical research. Different isoforms, alternative splicing, mutations, and posttranslational modifications to the troponins have been associated with the function and health of the heart as well as various pathologic conditions such as cardiomyopathies (Chap. 1).
Troponin C
Cardiac TnC is an 18 kDa EF-hand calcium-binding protein with two lobular domains, each of which has two Ca2+-binding sites. Each of the four EF-hands has two α-helices between which is a Ca2+-binding loop. There is an additional α-helix at the N-terminal (N-helix). The N-terminal binding sites control the regulatory function of the subunit. The same isoform of TnC is expressed in cardiac and slow skeletal muscle (ssTnC) and is not specific for the heart (c/ssTnC). Nonconservative amino acid substitutions in the first EF-hand of c/ss TnC impede calcium binding [13–15, 16, 18].
Troponin I
Cardiac TnI, a 24 kDa protein, was named for its inhibition of actin-activated myosin ATPase activity [22]. In humans, cardiac troponin I is produced from the TNNI3 gene on chromosome 19. When intracellular Ca2+ is low, the inhibitory region of cTnI binds to actin, inhibiting muscle contraction. Binding of Ca2+ to TnC increases as the intracellular concentration of Ca2+ increases. This induces a conformational change that increases the affinity of TnC for cTnI. Conformational changes to cTnI allow the molecule to alternate binding between TnC and actin in response to intracellular calcium concentrations [19]. Both cTnI and ssTnI are detectable throughout fetal development, but the skeletal isoform is predominant during that time period. There is a transition to expression of cTnI in the heart during the first nine postnatal months. Some data suggest that this isoform shift from fetal to adult may have functional implications for the heart. Hearts with congenital malformations may have a different time frame of isoform transition [20, 21].
Cardiac TnI has six distinct functional regions including: (1) an N-terminal extension, cTnI(1–30), unique to the cardiac isoform, containing phosphorylation sites Ser-23 and Ser-24; (2) the N-terminal, cTnI(34–71), a region that provides contact with troponin C; (3) cTnI(80–136) that binds to C-terminal regions of cTnT; (4) the overlapping inhibitory region, cTnI(128–147), that binds cTnC and actin-tropomyosin; (5) switch or triggering region, cTnI(147–163); and (6) the C-terminal domain that binds actin-tropomyosin, cTnI164-210 [17]. Residues 42–136 are sometimes referred to as part of the IT arm [18].
In addition to regulating the actin-myosin interaction, regions of the cTnI subunits as well as specific modifications to those regions have been shown to have additional regulatory control over cardiac function. The inhibitory region of cTnI contains Thr-143, a major phosphorylation site, which may affect velocity of shortening and is also implicated in the posttranslational modifications of cardiac troponins associated with hypertrophy and subsequent heart failure [22]. The phosphorylation sites in the NH2-terminal extension are targets for a number of protein kinases (PK), including protein kinase A (PKA). Under ß-adrenergic stimulation, the phosphorylation of Ser-23/Ser-24 decreases the Ca2+ affinity of the cTn complex and increases the rate of myocardial relaxation [23, 24]. Protein kinase C may phosphorylate cTnI at Ser-23/Ser-24, Ser-43/Ser-45, and Thr-144. Phosphorylation at Thr-144 appears to depress cooperative activation of the thin filament [25]. The process of dephosphorylation is little understood at this time. The N-terminal extension of cTnI may be removed, posttranslationally, due to hemodynamic stress or ß-adrenergic deficiency. This truncation removes the Ser 23/Ser-24 phosphorylation sites; however, the core structure of cTnI remains functional. Genetically modified mice with this alteration are viable through adulthood. The truncation of the N-terminal seems to increase the rate of relaxation of the ventricular muscle, increasing tolerance to decreased preload, an effect similar to phosphorylation of Ser 23/Ser-24 [24, 26–28].
Troponin T
Cardiac TnT, a 37 kDa protein, the largest of the three subunits, has diverse intermolecular interactions. Cardiac TnT binds to cTnI and cTnC to form the overall troponin complex and also binds to tropomyosin (Tm) and actin. In humans, three TnT genes have been described. These separate genes encode the isoforms found in slow skeletal muscle, fast skeletal muscle, and cardiac (cTnT) muscle. Each isoform is subject to alternative RNA splicing, producing multiple tissue-specific isoforms [19, 29]. The cardiac-specific TnT is encoded by the TNNT2 gene, located on chromosome 1. A number of isoforms have been identified, four of which have been described for the human heart. Cardiac TnT1 and TnT2 are expressed primarily in the fetal heart. Cardiac TnT3 is the primary isoform in the healthy adult heart, and cTnT4 is expressed in the fetal heart and is reexpressed in the adult failing heart. Although most of cTnT is highly conserved, there is a unique, hypervariable N-terminal region (residues 1–69) which is variable across isoforms [16, 19, 30].
Similar to cTnI, the N-terminal region of cTnT contains several phosphorylation sites, primarily Ser-2, Thr-294, Thr-204, Thr-213, and Ser-208. The role of cTnT phosphorylation and cardiac function is unclear, but data suggest that phosphorylation of cTnT decreases the binding affinity of cTnT for Tm and reduces the ability of troponin to expose blocked myosin binding sites. The net result is a decrease in Ca2+-dependent actomyosin ATPase activity [31]. A number of mutations associated with hypertrophic and dilated cardiomyopathies have been identified in human cTnT [32].
10.5 Cardiac Muscle Contraction
The sliding filament theory of muscle contraction was proposed in 1954 by Huxley and Hanson, years before it was recognized that calcium and the cardiac troponin complex played a key role in this process. The sliding of the interdigitated thin and thick filaments causes a shortening of the distance between the Z lines, as observed by microscopy. Later work demonstrated that contraction involves a cycle of myosin “reaching forward” to bind to actin and form the crossbridge, contraction, release of actin, and then the forward movement of the hinged globular region of the myosin S1 segment to begin the cycle again, as shown in Fig. 10.2.
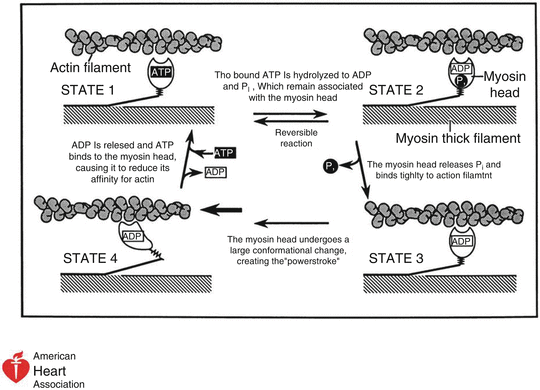
Fig. 10.2
Cardiac muscle contraction through one cycle of cardiac contraction and relaxation (This figure which originally appeared in Marian and Roberts [8] is used here with permission). In State 1, ATP is bound to the globular head of myosin. The myosin ATPase hydrolyzes ATP, generating ADP and Pi (State 2). The Pi is released and the globular head binds to the actin filament (State 3). Flexion of the hinge region of myosin displaces the globular head over the actin filament, causing the power stroke and muscle contraction (cardiac systole (State 3 to State 4)). ADP is released, with ATP again taken into the binding site on the globular head, releasing myosin from actin (return to State 1)
Hydrolysis of ATP provides the energy for myosin to pull the actin filaments. The myosin crossbridges rotate toward the center of the sarcomere in what is referred to as the “power stroke.” Subsequently, an ATP molecule binds to the myosin head, breaking the crossbridge and allowing myosin to reattach to the actin at a site further along the actin filament. After decades of rigorous scientific examination, the sliding filament model still stands, albeit refined by new data. A plethora of new proteins and lipids have been demonstrated to be involved in contraction, restoration of relaxation, and maintenance of structural integrity under conditions of consistent deformation (contraction-relaxation).
The cycle of a heartbeat is the process that leads to and includes contraction, ejection, subsequent relaxation, and filling of the cardiac chambers with blood. The cardiac cycle begins with an action potential and membrane depolarization. The concentration of intracellular Ca2+ rises, followed by Ca2+ binding to cTnC, changes in the thin filament, and force generation of crossbridges. The cycling of muscle fibers between contraction and relaxation must occur within the heartbeat and be adaptable to a wide range of heart rates and contractility. It has been proposed that the rise of intracellular Ca2+ and the Ca2+-induced troponin switch occurs within approximately 20 ms, the first 20–30 % of the isovolumetric pressure rise. This suggests that the rate of myocardial contraction is limited by processes other than Ca2+-regulated troponin switching [33] (Chap. 4).
In diastole, the N-terminal tail of cTnT, the inhibitory peptide of cTnI, and an actin-binding region of cTnI hold Tm in a blocking position that prevents the development of crossbridges (steric blocking model) [34]. The electrical events of the action potential and membrane depolarization are linked to the mechanical result of cardiac contraction. This excitation-contraction coupling begins with the release of calcium from the sarcoplasmic reticulum. Calcium enters the cytoplasm primarily via the L-type channels and triggers the release of more calcium from the sarcoplasmic reticulum via the ryanodine receptor (i.e., Ca2+-induced Ca2+ release). The increased level of cytosolic Ca2+ interacts with the regulatory N-domain of cTnC. The Ca2+ binding to cTnC induces exposure of a hydrophobic patch that draws the switch peptide region of cTnI to cTnC. The movement of the switch peptide toward the hydrophobic area of cTnC drags the adjoining inhibitory region of cTnI, causing release of the thin filament and an actin-binding site. The calcium-binding signal also induces release of the cTnT to tropomyosin interaction [5, 35]. The C-terminus of cTnI is carried along with this motion. The sequence is sometimes referred to as the “drag and release” mechanism [17]. This conformational change in the troponins allows actin crossbridge formation and sarcomeric shortening or contraction.
Return to the diastolic conformation of the cardiac troponin complex is an energy-requiring process that involves dissociation of calcium from cTnC, reversion of the conformation of the different subunits, and reduction of crossbridge formation. Four pathways modulate the removal of Ca2+ from the cytosol during the active process of relaxation, including the sarcoplasmic reticulum Ca2+ ATPase, sarcolemmal Na+/Ca2+ exchanger, mitochondrial Ca2+ uniporter, and sarcolemmal Ca2+ ATPase [35]. Factors such as phosphorylation of the subunits, mutations, and isoform switching of the troponin subunits as well as the assay system used (e.g., isolated subunit versus troponin complex, reconstituted into thin filament or cardiac myocyte) can alter the kinetics of Ca2+ association and dissociation with cTnC [23]. Liu evaluated the effect of cTnI and cTnT modifications associated with cardiac disease on the rates of calcium association and dissociation both in isolated troponin complex and when these modifications were reconstituted into the thin filament [36]. Effects on the kinetics of Ca2+ association and dissociation from the reconstituted thin filament were observed when almost no effects were detected in the isolated cTn complex. The majority of disease-related protein modifications did not alter the Ca2+-binding properties of the isolated troponin complex. However, when reconstituted into the thin filament, the mutations associated with dilated cardiomyopathy decreased the Ca2+ sensitivity of the thin filament. The mutations associated with restrictive and hypertrophic cardiomyopathy, as well as the ischemia-induced truncation of cTnI, increased the Ca2+ sensitivity of the thin filament. The various protein modifications altered the steady-state Ca2+ binding to TnC by influencing both the Ca2+ association and dissociation rates on the thin filament.
10.6 Increases in Circulating Cardiac Troponins
The circulating forms of cTns (i.e., those proteins identified in the systemic circulation after a myocardial infarction) include cTnT, cTnI, the cTnI-TnC complex, and a ternary complex of cTnT-cTnI-TnC [37, 47]. Most cTns are present in the myocyte as part of the structural elements. An estimated 3–8 % of the total cellular troponin is free in the cytoplasmic pool. It is hypothesized that free troponin in the cytoplasmic pool contributes to initial increases in circulating cTn following myocardial injury. Subsequent increases are thought to be due to degradation of structural proteins [38]. The probability of proteins the size of the troponins leaking from a viable cell is uncertain. The leakage from viable cells of fragments detectable by assays is possible but raises additional questions.
There are six proposed mechanisms for cTn release: (1) myocyte necrosis, (2) apoptosis, (3) normal myocyte cell turnover, (4) cellular release of proteolytic troponin degradation products, (5) increased cellular wall permeability, and (6) formation and release of membranous blebs. Cardiac myocyte death can be further categorized to include pathways of extrinsic death receptor pathway, intrinsic apoptosis, necrosis, necroptosis, and possibly others [39, 40]. How the mechanism of damage influences the form in which the troponins are released is not completely characterized.
Some baseline level of circulating cTns might be expected due to normal maintenance and turnover of cardiac myocytes. Replacement of cTns includes the synthesis, assembly, and degradation of the individual components of the cardiac sarcomere. The mechanism by which the individual components of this multi-protein complex are removed and replaced while maintaining the functionality of the contractile unit is incompletely understood. In rats, the rate of incorporation of radiolabeled amino acids into the troponin complex in situ demonstrated that cTnI and cTnT had similar half-lives (i.e., similar turnover rate) of 3.2 and 3.5 days, respectively. These half-lives were significantly different from cTnC’s half-life of 5.3 days [41].
It is generally accepted that circulating cTn is highly susceptible to phosphorylation and proteolysis. Regardless of the mechanism by which troponins are released into circulation, some sections of the complex and fragments are more susceptible than others to degradation and modification. Modifications may include acetylation, protein sequence variants such as mutants, alternatively spliced isoforms, amino acid polymorphisms, and protein complexes. Each of these posttranslational modifications may have variable effects on the ability of a given antibody to detect cTnI [42]. Degradation of cTnT after release into the circulation is not as well understood. At least in hemodialysis patients, circulating cTnT molecules are degraded into smaller fragments that may be detectable by assays [43]. The process and overall kinetics of clearance are incompletely understood.
10.7 Assays
In 1987, Cummins et al. [58] described a radioimmunoassay for cardiac troponin I. Two years later, Katus et al. [59] announced an enzyme-linked immunosorbent assay (ELISA) for cardiac troponin T. Currently, there is one commercially available cTnT assay and numerous commercially available cTnI assays.
In this chapter, assays will be discussed in terms of sensitivity, specificity, and factors that may influence detection. The diagnostic interpretation of cardiac troponin increases and the clinical application are discussed in the next Chapter.
The oldest category of commercial assays, no longer in use, was less sensitive in detecting increases in cTn, compared to newer assays. A 10 % coefficient of variation was achieved at approximately 1 μg/l, and, therefore, pathologic elevations were detected almost exclusively. The current assays have third- and fourth-generation antibodies and incorporate newer technology to increase the analytical sensitivity. Assays currently available are informally referred to as high-sensitive, ultrasensitive, or sensitive-contemporary assays. These assays reliably detect to greater than the 99th percentile value but only quantitate cTn in a fraction of clinically healthy individuals. The IFCC recommends describing an assay as “high sensitivity” only if the cTn is measurable in more than 50 % of healthy subjects (and preferably in more than 95 %) below the 99th percentile of the assay and above the assay’s limit of detection. The total imprecision (coefficient of variation) at the 99th percentile should be ≤10 % [45, 46]. Conrad and Jarolim suggest reserving the term “ultrasensitive” for assays capable of quantitating cTn at levels less than the lowest cTn concentrations seen in healthy individuals [46].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


