Figure 25–1.
(a) Electrocardiographic (top) and vectorcardiographic (bottom) representation of a long-term CM development in a chronic canine model. Leads I, aVF, and frontal plane VCG shown at baseline, during ventricular pacing, 1 h after ventricular pacing for 7, 21 days, and 3 days after cessation of ventricular pacing. Note the increase in T vector amplitude and rotation towards the direction of the paced QRS complex (arrows). Cross-hair represents calibration at 1 mV (From Yu et al. [31]. Reprinted with permission from Wolters Kluwer Health). (b) Quantitative assessment of CM. The control and memory T vector loops superimposed. CM can be measured by the change in T vector direction, amplitude, and 3-dimensional distance (T peak displacement, TPD measured in mV) between T vector peaks (“T control” represents the baseline T vector loop, “T memory” represents T vector loop after CM induction). TPD is the preferred method of CM measurement (From Plotnikov et al. [27]. Reprinted with permission from Oxford University Press)
A vectorcardiographic-based method of CM measurement facilitated quantitative assessment of T wave changes independent of the individual ECG lead characteristics. It involves measuring a 3-dimensional distance (in mV) between the T vector peaks at baseline and after the induction of CM [27], Fig. 25.1b. The proposed measurement (T peak distance, TPD) has become used in CM quantification in both animal and human studies [33, 40–42].
Although prolonged abnormal ventricular activation eventually results in structural changes in the left ventricle regardless of its mechanism [43–45], the ventricular pacing necessary for the development of the long term CM was shown to produce no changes in myocardial blood flow, cell size, and no histological evidence of hypertrophy, ischemia, or necrosis [1, 25, 31]. Therefore, CM can be considered an early, purely electrical stage of the long-term ventricular remodeling that can eventually progress to asymmetric ventricular hypertrophy, dilatation, and heart failure [39, 44, 45].
Molecular Mechanisms of CM
The first and most extensively studied ion channel implicated in the development of short-term CM was Ito. In an acute open-chest canine model Del Balzo et al. used 4-aminopyridine (4-AP), which blocks Ito blocker to abolish the T wave changes induced by 20-min ventricular pacing [26]. Based on the preferential expression of Ito in canine epicardium [46] the mechanism of short-term CM was proposed to include a change in transmural voltage repolarization gradient. Further support for this hypothesis was found by studying action potentials in isolated endocardial and epicardial slabs while simulating induction of CM by pacing in the direction perpendicular to the fiber direction. This intervention abolished phase 1 notch, prolonged the action potential of the epicardial cells, and reduced the transmural repolarization gradient. Pretreatment of the slabs with 4-AP reduced the phase 1 notch in the epicardial tissue and no further changes in action potential shape and duration were observed during pacing [47].
In long-term CM experiments prolongation of the epicardial action potential duration [25], loss of the phase 1 notch, decrease in Ito density, and decreased mRNA levels for Kv4.3, the gene controlling the pore-forming unit of the channel carrying canine Ito [31] and in the protein of its accessory subunit, KChIP2 [29] were observed in the left ventricular epicardium, indicating a role for Ito in the evolution of long-term CM as well (Fig. 25.2).
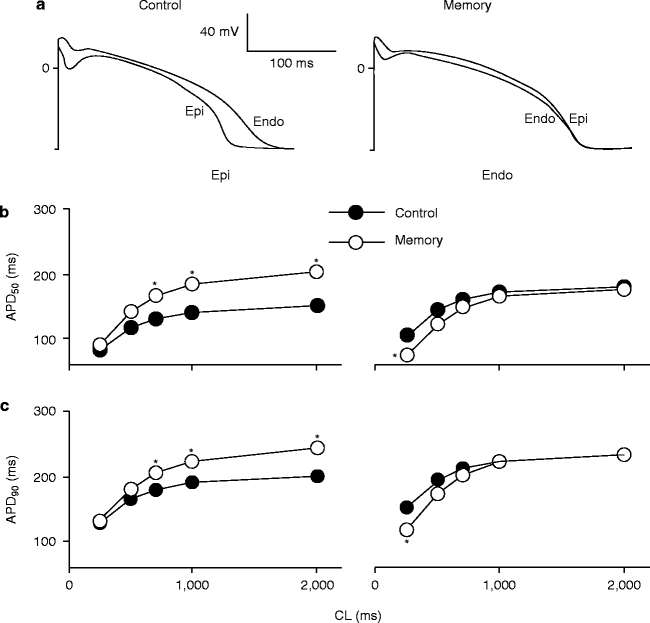
Figure 25–2.
CM-induced changes in the action potential shape and duration. (a) Representative experiments showing superimposed epicardial and endocardial AP in a control dog and a dog with CM at CL = 1,000 ms. (b, c) Effects of CM on APD50 (b) and APD 90 (c) in epicardium and endocardium. *P < 0.05 vs respective control a the same CL (From Yu et al. (31) Reprinted with permission from Elsevier).
While the direction of changes in Ito density and distribution is similar in short- and long-term CM, their mechanisms appear to be different. In short-term CM the decrease in Ito is believed to result from internalization of the AT1 angiotensin receptor/Kv4.3/KchIP2 complex (discussed further below) [48]. In long-term CM it is achieved by the reduction in the transcription factor CREB (cAMP response element binding protein). Critical to this observation is a cyclic AMP response element near the promoter region of KChIP2 and Kv4.3. A reduction in CREB would lead to decreased synthesis of the channel components. Both the short- and long-term processes appear to be initiated by angiotensin II synthesis and release and may involve the actions of reactive oxygen species [29].
Several additional ion channels are implicated in short- and long-term CM including IKr [32], ICa,L [24], possibly by the mechanism of CREB affecting transcription of some of them [29]. The reversal of the normal epicardial > endocardial gradient for the IKr density and the loss of the gradient for ERG mRNA and protein were documented in long-term CM around the ventricular pacing site also contributing to the local epicardial APD prolongation [32]. Reduction in epicardial connexin43 expression close to the pacing site (but not at remote ventricular areas) with gap junction redistribution was also observed along with the decrease in the conduction velocity around the pacing site [49] and an increase in the activation time during normal conduction [33] in long-term CM. A schematic of the short- and long-term CM development is presented in Fig. 25.3.
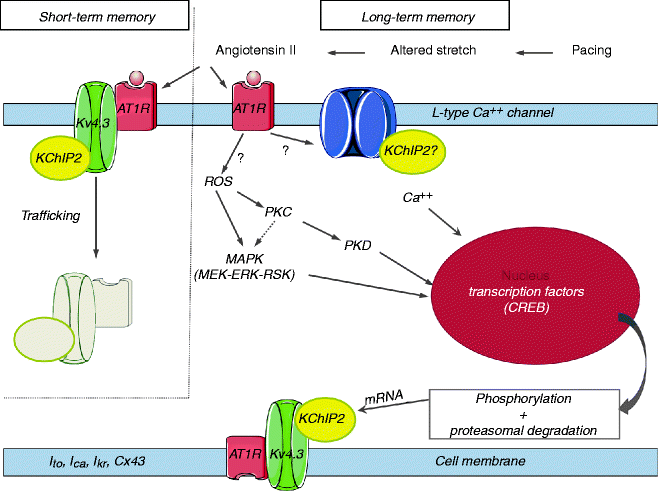
Figure 25–3.
Schematic of pathways already identified and under study in short-term and long-term cardiac memory. Aberrant activation alters stretch resulting in increased cardiac angiotensin II synthesis/release. Short-term memory results from angiotensin II-induced trafficking of an AT1 receptor/Kv4.3/KChIP2 macromolecular complex. The result is reduction of Ito. Long-term memory is initiated by angiotensin II binding to its receptor leading to reduction of the transcription factor CREB. Production of reactive oxygen species (ROS) leading to MAPkinase and protein kinase C/D-determined pathways are possilbe intermediary steps. CREB is phosphorylated and then proteasomally degraded, resulting in reduced KChIP2 transcription and expression and decreased Ito. Other ion currents altered in memory include ICa, L and IKr. Connexin43 is also involved. Ca2+ is an important cofactor throughout (From Ozgen and Rosen [23]. Reprinted with permission from Elsevier)
It is important to point out that the majority of the biochemical studies in CM involved tissue samples obtained from the areas 1–2 cm away from the left ventricular pacing site and although a gradient change in the transcription pathways across the ventricle was documented [34] the regional differences (especially in relation to the local mechanical function) have not been fully explored.
The processes of ventricular remodeling operate in the different directions in early- and late-contracting areas. The late results of this differential remodeling of the ventricle are quite opposite: thinning and atrophy of the early-contracting areas and hypertrophy of the late-contracting regions [44, 45]. Therefore it is plausible to predict that different mechanisms of remodeling operate in different areas of the ventricle at earlier stages as well which requires further investigation.
CM Triggers: Mechanical Forces Versus Electrical Activation Sequence
Along with the signal transduction and molecular control of CM its triggering factors have been extensively studied. It was clear that changes on a cellular and molecular level cannot be directly explained by the change in electrical activation sequence itself (a single cell cannot possess the knowledge of the timing of the activation front); therefore other mechanisms have to be involved. From the early experiments the alteration in the local stress-strain relationship and geometry of ventricular contraction triggering activation of the renin-angiotensin system was suspected as the CM trigger and confirmed experimentally. Pretreatment of animals with saralasin, (AT1 and AT2 receptor antagonist), captopril (angiotensin converting enzyme inhibitor), and chymostatin (inhibitor of tissue chymase, which converts angiotensin I to angiotensin II) abolished the development of short-term CM suggesting a role for local angiotensin II production in its initiation [50].
Additional proof of local angiotensin II release as the CM trigger came from the demonstration that AT1 receptors co-localize with the α (alpha) (Kv4.3) and accessory (KChIP2) subunits of the Ito channel. Exposure to angiotensin II caused internalization of the AT1R–Kv4.3-KChIP2 macromolecular complex [48] reducing Ito current in HEK cells in which the channel components of the complex were overexpressed. The results were validated in co-immunoprecipitation experiments in cardiac myocytes, and in experiments using FRET. Another mechanism by which altered strain triggers CM is the stretch activated receptors (SAR) [2, 40]. In a canine model SAR blockade with streptomycin dramatically attenuated CM [40].
Sosunov et al. [51] in an isolated perfused rabbit heart obtained direct evidence that it is indeed mechanical factors and not the electrical activation sequence itself that serves as the primary CM trigger. The excitation-contraction uncoupler blebbistatin completely abolished CM after ventricular pacing, while local myocardial stretch without ventricular pacing (and therefore unchanged activation) produced T wave changes similar to CM. This observation suggests potential common mechanisms of T wave changes observed in CM and various forms of structural heart disease associated with mechanical dyssynchrony without significant QRS prolongation such as myocardial infarction, hypertrophy, certain cardiomyopathies, etc.
Electrophysiological Effects of Cardiac Memory
While the studies of molecular mechanisms associated with CM in a large part have reached consensus, the electrophysiological effects of CM at the level of the whole heart are less clear.
There is an ongoing debate regarding to what degree transmural vs regional repolarization gradients are responsible for the T wave changes in CM. In perfused myocardial wedge preparations Jeyaraj et al. found APD prolongation in late- (and, to lesser extent early-activated regions) suggesting that regional repolarization gradient is responsible for long-term CM, while small transmural repolarization gradient remained unchanged [1]. In contrast, Spragg et al. using ablation-induced LBBB demonstrated shortening of action potential duration, effective refractory periods, and conduction velocity in the wedge preparations from the late-activated areas with increased tissue strain [2]. APD shortening in the late-activated areas was also consistently observed in the isolated rabbit heart [19, 51]. In the intact in-situ canine heart Coronel et al. showed the development of the transmural repolarization gradient localized at the area close to the pacing site with no evidence for significant regional gradient during long-term CM [33]. In-situ study of the short-term CM by the same group showed baseline apical-basal repolarization gradient that did not change significantly although repolarization time shortened uniformly in the setting of short-term CM [30].
Using non-invasive ECG imaging (ECGI) in patients with WPW syndrome before and after ablation of an accessory pathway Ghosh et al. detected the presence of a large (85–95 ms) regional repolarization gradient between the accessory pathway ventricular insertion site and the rest of the myocardium (translating into apical-basal gradient given the annular location of the accessory pathways). This gradient was caused by a significantly prolonged activation-recovery time in the area of the bypass tract insertion that gradually resolved within 1 month consistent with the time course of the long-term CM [52].
The challenges observed in extrapolating the results obtained in isolated myocytes, tissue slabs, and wedge preparations to the effects of CM in the heart in situ mirror those in explaining the genesis of the normal T wave [53, 54] (see [55] for potential explanations and discussion).
The relevance of these findings to human CM (the major contributors to which are right ventricular pacing and LBBB) is unclear as well. The vast majority of animal CM models followed the original idea of complete reversal of left ventricular activation by stimulating the latest activated site during normal conduction (i.e. posterolateral LV base) [36, 37]. However the degree of mechanical dyssynchrony which (rather than the electrical conduction delay) is the primary CM trigger does not correspond to the degree of activation reversal. Instead, endocardial right ventricular apical (rather than the epicardial left ventricular basal) pacing was found to create the most dyssynchronous contraction [39, 43]. This is one of the possible reasons why CM following right ventricular pacing in humans achieves the steady state more rapidly (1 week) [56, 57] than left ventricular pacing.
Human Studies of Cardiac Memory
Time Course of CM Development
Unlike the majority of animal experiments most human data on CM were obtained using right ventricular apical endocardial pacing. Compared to the long-term animal models, CM in humans evolves faster, achieving steady state within a week of right ventricular pacing at physiologic rates when assessed semi-quantitatively [56, 57]. However, after 1 week of ventricular pacing T vector during normal conduction still has not been fully aligned with a paced QRS [58] and T vector loop morphology continued to evolve [57] suggesting that further subtle changes in T vector likely to occur beyond this period. In a single patient with experimentally induced rate-related LBBB M. Rosenbaum observed apparent plateau in ECG changes as shortly as after 24 h of abnormal conduction [17] although in cases of rate-dependent block pre-existing memory cannot be ruled out.
Effects on Myocardial Repolarization
Several studies documented the opposite effects of CM in humans on repolarization during normal and aberrant conduction. While during normal conduction (sinus rhythm or atrial pacing with narrow QRS) QT interval prolongs in the setting of long-term CM by 4–5 %, QT interval during ventricular pacing shortens with the increased duration of pacing by approximately 9 % [57–59]. This conforms to the concept that CM is an adaptive reaction to the new activation sequence [19, 51]. The fact that in the setting of CM changes in ventricular activation sequence (switching from sinus rhythm to ventricular pacing and back) produce instantaneous contrasting changes in the duration of repolarization (shortening with ventricular pacing and prolongation in sinus rhythm) indicate that CM involves a complex change in both transmural and regional repolarization gradients. Changes in T vector loop morphology that could be consistent with pro-arrhythmogenic effect during normal activation were observed [57].
It is unclear whether these changes result in untoward clinical consequences. An isolated case report of pro-arrhythmic effect of CM after ablation of an accessory pathway [60] was confounded by an iatrogenic right bundle branch block and therefore is questionable. Although long-term CM affects the action of antiarrhythmic drugs (for example exaggerating QT-prolonging effects of IKr blockers) in animals [27], the clinical significance of it is unclear.
Effects on Contractile Function
Little is known regarding contractile function of the heart and CM in humans. Alessandrini et al. [59] demonstrated that long-term CM is associated with transient diastolic dysfunction (prolongation of the isovolumic ventricular relaxation time and decrease in the left ventricular filling rate, dD/dt/D). The causality of the relation to CM was proven by correlation of the magnitude of diastolic dysfunction to the change in QT interval and T wave amplitude. These changes in myocardial function resolved within 1 h after cessation of ventricular pacing.
Longer-lasting myocardial systolic dysfunction (a decrease in the LV ejection fraction lasting over 24 h) was observed after 1 week of ventricular pacing [61].
Little is known regarding CM in structural heart disease besides a small observation series in hypertrophic obstructive cardiomyopathy [62]. Patients with compromised baseline ventricular function are more likely to develop pacing-induced ventricular dysfunction and heart failure [63, 64]. How exactly the phenomenon of CM relates to the pacing-induced heart failure in patients with compromised systolic function is unknown.
Clinical Implications of CM
Differential Diagnosis Between CM and Ischemia
Since the time CM was first recognized its similarity to ischemic T wave inversions (TWI) made it a significant confounder in the diagnosis of myocardial ischemia. This diagnostic dilemma becomes even more important because the pattern of the precordial TWI due to CM can closely resemble the pattern seen in proximal left anterior descending artery (LAD) disease (so called Wellens’ syndrome) which portends poor prognosis unless urgent revascularization is performed (Fig. 25.4) [65, 66]. These changes are believed to be due to a transient LAD occlusion that resolves spontaneously and at the time of presentation these patients are often asymptomatic, further complicating the diagnosis [66]. As the result, asymptomatic precordial TWI is a common referral reason for cardiac catheterization.
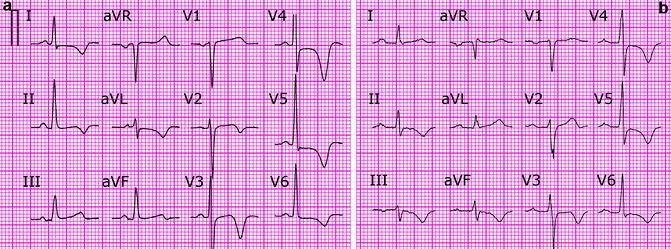
Figure 25–4.
(a) Precordial T wave inversions (TWI) in a patient with transient proximal LAD occlusion (Wellens’ syndrome). TWI in leads I, aVL is present. (b) Precordial T wave inversions due to CM. Note positive T waves in leads I, aVL and TWI V4–5 > TWI III
In developing diagnostic criteria to distinguish between TWI due to ischemia and CM due to intermittent ventricular pacing we used the following premises:
1.
The direction of the ischemic T waves points away from the area of ischemia. Since in most cases the ischemic region involves the left ventricle, the ischemic TWI should have a rightward axis in the frontal plane. Repolarization abnormalities due to anterolateral ischemia in particular are characterized by TWI in leads I and aVL.
2.
The vast majority of pacemaker leads are implanted in or close to the right ventricular apex producing left superior QRS axis during ventricular pacing with QRS positive in leads I and aVL. Pacing from alternative right ventricular sites (such as septal inflow tract and high interventricular septum close to the His bundle) usually also produce positive QRS complex in these leads during pacing. Based on the principles of CM this should result in positive T waves in these leads upon resumption of normal conduction.
3.
Maximal repolarization changes due to CM occur in two non-contiguous areas (the earliest and latest activation sites) which is likely to produce a unique direction of T wave vector that cannot be replicated by repolarization changes in any single territory.
Based on these premises we compared in a retrospective study the direction of the TWI caused by ischemia and CM [67]. As predicted, the frontal plane CM-induced TWI direction was quite different from ischemia in the LAD and left circumflex artery (LCX) distributions. When translated to the 12-lead ECG terms, the presence of positive/isoelectric T wave in lead I and positive T wave in lead aVL while typical for CM was never seen in LAD/LCX ischemia. It was observed in some cases of right coronary artery (RCA) ischemia; however those cases differ from CM-induced TWI in other vector planes. When translated to the 12-lead ECG terms this pattern was characterized by TWI that was deeper in the inferior leads (II, III, aVF) compared to the deepest TWI in the precordial leads whereas the reverse was true for CM-induced TWI. Overall, the combination of the two criteria:
1.
Positive/isoelectric T lead I and positive T aVL and
2.
Precordial TWI > TWI in lead III was 92 % sensitive and 100 % specific for CM in discriminating it from ischemia.
Although not confirmed in larger prospective trials, these criteria have been used successfully in clinical practice by us as well as others [68, 69]. Since the axis during right ventricular pacing and left bundle branch block (LBBB) is similar with positive QRS complex in leads I and aVL, the criteria described can also be used in the cases of TWI due to intermittent LBBB [69].
One of the limitations in using this diagnostic tool is the concern that T wave changes due to CM can counter-balance or obscure ischemic TWI when CM and ischemia occur together in the same patient. Although CM has been studied in populations of patients with structural heart disease such as hypertrophic obstructive cardiomyopathy [62], no published data are available in patients with T wave abnormalities due to coronary artery disease. Our preliminary results in this population indicate that CM does not “over-ride” ischemic TWI eliminating the possibility of missing myocardial ischemia. However, when TWI in leads I and aVL are present at baseline as the result of the structural heart disease, T waves stay inverted despite the development of CM (T vector never fully aligns with the paced QRS vector) which can result in over-diagnosis of ischemia.
CM During Continuous Abnormal Ventricular Activation
Since the introduction of the original concept of cardiac memory in 1982 and until recently, two important issues remained unresolved.
First, it was universally accepted that the return to the normal ventricular activation sequence is pre-requisite to observe it. Although it was clear that CM develops gradually during the period of abnormal activation secondary T wave changes were thought to completely obscure it.
The most common causes of CM in clinical practice – LBBB and atrioventricular block requiring ventricular pacing are rarely reversible. This significantly limited clinical application of CM as the majority of the patients developing CM never return to normal conduction.
Second, although obvious individual variations in the degree of CM, were observed [70], it was unclear what factors determine this variability. The only factor associated with CM magnitude was the amount of ventricular pacing [57].
Using further refinement of the quantitative methods of CM assessment developed previously in the animal studies [27], we sought to resolve these issues.
In our clinical practice we observed that following pacemaker implantation the amplitude of T waves during ventricular pacing decreases in time. We hypothesized that this change in T wave amplitude is related to the development of CM. Using vectorcardiographic software [71] we compared 3 dimensional T vector loop displacement after 1 week of ventricular pacing (DDD with short atrioventricular delay) in humans in AAI mode (conventional measure of CM) to that in DDD mode with continuous ventricular pacing [58].
We found that not only the 3-dimensional direction of the T wave shift was identical when measured in AAI and DDD mode (Fig. 25.5), but the magnitude of the shift in the two pacing modes correlated closely (Fig. 25.6). In contrast to AAI mode (conventional CM) which demonstrated typical rotation of the T wave vector towards the paced QRS direction, CM in DDD mode (continuous ventricular pacing) was characterized by a progressive decrease in discordant T vector magnitude without change in direction. The strong correlation between T vector changes in both pacing modes suggested that they reflect the same physiological phenomenon. These findings have important implications.
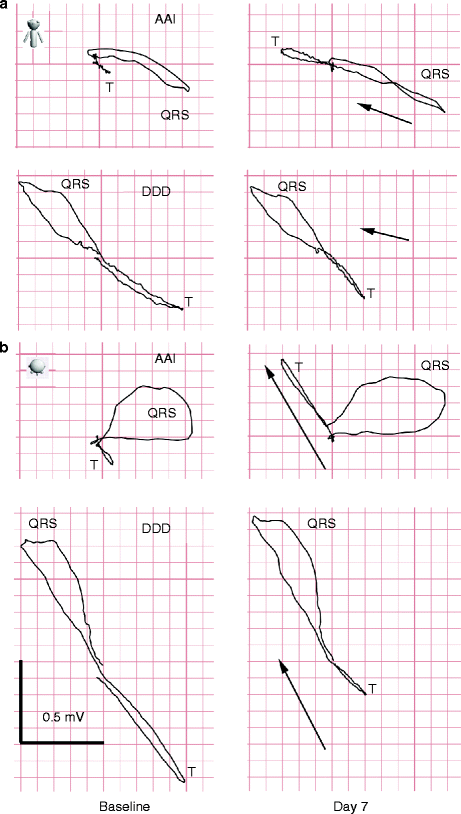
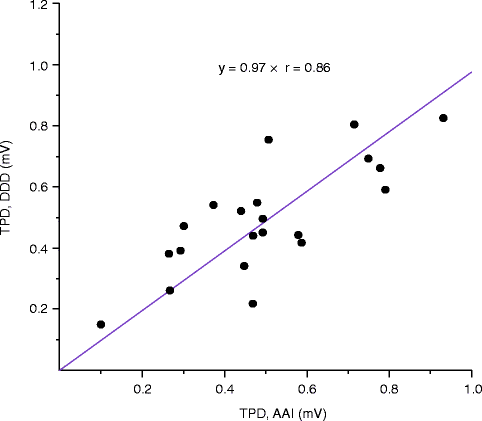
Figure 25–5.
Vectorcardiogram of the same patient during AAI and DDD pacing in frontal (a) and transverse (b) projections before (baseline) and after induction of cardiac memory (Day 7). On Day 7 during AAI pacing, the T vector assumes the direction of the paced QRS complex while increasing in magnitude. At the same time in DDD mode, the T-vector magnitude decreases without significant change in direction. Black arrows indicate the direction and magnitude of the projection of T-peak displacement (TPD) as the result of cardiac memory and are very similar in AAI and DDD modes. Sagittal plane (not shown) showed similar changes (From Shvilkin et al. [58]. Reprinted with permission from Elsevier)
Figure 25–6.




CM magnitude (T peak displacement, TPD) measured during normal (AAI pacing) and aberrant (DDD pacing) ventricular activation. CM magnitude measured in the two pacing modes correlates closely (From Shvilkin et al. [58]. Reprinted with permission from Elsevier)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
