Figure 16–1.
The structural basis of KATP channels. (a) KATP channels are formed from Kir6 (left) and SUR (right) subunits. Kir6 subunits each consist of two transmembrane helices (M1, M2), a pore-forming region (including the P-helix and selectivity filter), and cytoplasmic NH2 terminus (including the amphipathic slide helix) and COOH terminus. SUR consists of the TM0 and L0 regions that interact with and modulate gating of Kir6, followed by two additional 6-helix TM regions (TM1 and TM2), each followed by a nucleotide binding fold (NBF). The two nucleotide binding folds together generate two ATP/ADP binding sites (ABS1 and ABS2) at their interface. (b) The KATP channel is a heteroctamer of four Kir6 subunits which form the central pore with each Kir6 associated with one SUR subunit. (c) Homology models, based on crystal structures of related proteins, predict one inhibitory ATP binding site at each of the Kir6.x interfaces and Mg-dependent ATP/ADP binding sites (ABSs) in the dimeric nucleotide binding folds (NBF) of SUR. (d) Human SUR and Kir6 gene structures indicate that each pair is located adjacent to each other on two chromosomes
Chamber-Specificity of Cardiac KATP Channels
For many years, there was a consensus that cardiac KATP channels were essentially exclusively formed by Kir6.2/SUR2A complexes. Heterologously expressed Kir6.2 and SUR2A recapitulate essential cardiac ventricular KATP channel properties [29, 32], and Kir6.2 and SUR2A functionally [32] and physically [33] interact. There is also an absence of KATP channel activity in the ventricles of Kir6.2 knockout (Kir6.2−/−) mice [34] and disruption of the SUR2 gene greatly reduces sarcolemmal KATP channel activity [35, 36]. Normal activity is reported in ventricular myocytes from both Kir6.1 knockout (Kir6.1−/−) [37] mice and SUR1 knockout (SUR1−/−) mice [38].
However, there is also evidence that all Kir subunits (Kir6.1 and Kir6.2) and SUR subunits (SUR1 and SUR2A/B) are expressed in the heart and play a functional role [15, 16, 18, 29, 39–41]. Results from heterologous expression systems show that more than one type of Kir6.x or SURx subunit can exist within a single functional channel [42–46], suggesting that the native composition of each subunit might be heterogeneous. The first hint of a role for Kir6.1 in cardiomyocytes was that a dominant negative Kir6.1 suppressed cardiac KATP [40], although others have not seen a comparable effect [47]. Additionally, a gain-of-function mutation in Kir6.1 has been linked to Early Repolarization Syndrome [6–8], although the specific cell type in which the mutation might be acting is unknown.
Confirming a role for SUR1 in the heart, a recent study has shown that KATP currents are abolished in atrial myocytes from SUR1−/− mice while remaining unchanged in ventricles [38].
Pharmacological studies show that the atrial KATP channel is more sensitive to diazoxide than pinacidil, indicating that the Kir6.2/SUR1 complex is present. Conversely, the ventricular KATP channel is more sensitive to pinacidil than diazoxide implying the presence of the Kir6.2/SUR2A complex [38]. This notion is further supported by a subsequent study on intact hearts [48] in which only diazoxide reduces action potential duration (APD) in atria while only pinacidil reduces APD in ventricles. Moreover, the diazoxide effect is abolished in atria from SUR1−/− mice while pinacidil action is unaffected in ventricles [48]. These studies demonstrate a previously unappreciated chamber-specific KATP channel heterogeneity in the heart. Though the functional consequences of having different SUR subunits in atria and ventricles is not yet well understood, the presence of the SUR1 subunit in atria may have clinical consequences since SUR1 is the most sensitive subtype to changes in nucleotide concentrations [49], as well as a differential pharmacological sensitivity [50, 51].
KATP Channels in the Cardiac Conduction System
KATP channels are also present in the cardiac conduction system. Electrophysiological studies have detected KATP channels in rabbit sinoatrial node (SAN) cells [52], atrioventicular node (AVN) cells [53], and Purkinje cells [54]. The reported properties of these KATP channels in the conduction system are distinct from those in ventricular myocytes, suggesting a different KATP channel composition. Specifically, these channels have a smaller single-channel conductance, when recorded in rabbit SAN cells (50 ps) [52] and in Purkinje cells (60 ps) [54] as well as in mouse cardiac conductance system (57 ps) [55] than that in ventricular myocytes (80 ps), which would point to Kir6.1 with its smaller conductance. A recent study revealed Kir6.1, Kir6.2, and SUR2 RNA messages in mouse SAN and AVN tissue as well as atria and ventricles [56]. However, KATP currents are abolished in SAN cells from Kir6.2−/− mice, which indicates that Kir6.2 is an essential subunit in the SAN [57]. In the same study, sarcolemmal KATP channel activation inhibited SAN automaticity during hypoxia which indicates a potential role for sarcolemmal KATP channels in regulating heart rhythm during pathological conditions. Glibenclamide, a selective KATP channel antagonist, prevents cardiac conduction delay caused by low-flow ischemia in perfused mouse hearts [55], indicating a role of KATP channels in modulating cardiac conduction under metabolic stress. Computer simulations predict that activation of KATP channels by 1–5 mM cytoplasmic ATP will cause significant shortening of the Purkinje cell action potential [54], suggesting that KATP channels may have an important influence on atrioventricular conduction in response to changes in heart metabolism.
In conclusion, cardiac KATP channels are not exclusively generated by Kir6.2/SUR2A complexes nor are the different isoforms uniformly distributed through the heart. At least in mice, atrial KATP channels are formed by the Kir6.2/SUR1 complex. KATP composition in other species has not yet been systematically studied, but pharmacological studies in human hearts suggest that SUR1 and SUR2A may be both present, more interestingly, remodeling in molecular composition may happen in heart disease condition [58]. In addition, Kir6.1 may play a critical role in the cardiac conduction system. This diversity of subunit composition within the heart may allow KATP channels to function in distinct functional roles depending on the cell type in which they are expressed.
Regulation of Cardiac KATP Channels
Regulation by Nucleotides
Cytoplasmic ATP inhibits cardiac KATP channels with a half-maximal inhibition (K1/2) of ∼10–50 μM [59–63]. Recognition of the heteromultimeric architecture of the channel and molecular cloning of the cDNA encoding each subunit allowed the identification of amino acid residues that determine ATP inhibition. Pore-forming Kir6.2 subunits alone, without regulatory SUR subunits do not effectively traffic to the plasma membrane [64–67]. However, truncated Kir6.2, lacking the C-terminal RKR endoplasmic reticulum retention signal, can reach the membrane [64]. These truncated Kir6.2 generated channels retain ATP-induced inhibition, implying that the determinants are contained within the Kir6 subunit [64]. The cytoplasmic N- and C-termini of Kir6 form an ATP binding pocket with a total of four binding pockets per channel [68–72] at the interface of adjacent subunits (see Fig. 16.1c). Phosphorylation is not necessary for channel inhibition because nonhydrolyzable analogues of ATP are still effective [60, 73, 74]. In the absence of Mg2+, reducing the number of phosphate groups from three to two (ADP) or one (AMP) also blocks channel activity but with reduced affinity [59, 60, 75–77], suggesting an important role of the γ-phosphate for ATP binding and KATP channel inhibition.
In the presence of Mg2+, both ATP and ADP stimulate channel activity [14, 60, 63, 78–81]. Mg-nucleotide stimulation and responsiveness to diazoxide and sulfonylurea require expression with SUR subunits [15, 29, 51, 64, 82–86], indicating that the structural elements responsible for MgADP stimulation and other KATP modulators are on the SUR subunit. Mutations of key SUR subunit residues disrupt the response of KATP channels to MgATP and MgADP [87, 88], as well as KATP channel openers [82, 89]. The nucleotide binding domains-NBD1 and NBD2, located between the eleventh and twelfth transmembrane regions and the C-terminus of SUR subunit, are essential structures for nucleotide binding and stimulation [82, 89–92] (see Fig. 16.1a, c). Mutations in NBD1 prevent ATP binding at both NBDs and interfere KATP channel openers acting on NBD2 [82, 88, 89, 93, 94]. SUR1 and 2A have intrinsic Mg2+-dependent ATPase activity which hydrolyzes ATP at the NBDs, in particular NBD2 [95–97], and evokes a “power stroke” which overcomes ATP inhibition of Kir6 [97, 98]. Experimentally, MgADP stimulation is more effective than MgATP [14, 60, 63, 78–81] suggesting that binding of MgADP at the NBDs maintains an activated state of SUR, reducing Kir6.2 sensitivity to ATP inhibition [14, 82, 98, 99]. Binding of MgADP at NBD2 may also promote ATP binding at NBD1 [91, 98, 100]. Although there are currently no crystal structures of SUR subunits or domains, related bacterial NBDs crystallize as head-to-tail dimmers [101], with two ATP binding sites (ABSs) formed at the dimer interface. A recent mutagenesis study indicates that both NBDs form as a heterodimer and mutations at the dimer interface disrupt stimulation by MgADP and diazoxide, suggesting an important role for the NBDs in SUR-dependent regulation [101].
Regulation by Anionic Lipids
Membrane phosphoinositides potently stimulate KATP activity by interaction with the Kir6.2 subunit [102–106]. Application of phosphatidylinositol 4,5-bisphosphate (PIP2) to the cytoplasmic membrane increases channel open probability and decreases ATP sensitivity [102–104, 107], demonstrating that ATP inhibition is a dynamic property that is dependent on membrane composition. PIP2 and ATP bind competitively to KATP channels [105, 108]. However, this apparent competition may be revealing allosteric regulation instead of competitive ligands [72, 109]. Following the activation of KATP channels during metabolic inhibition, a secondary decrease in PIP and PIP2 causes the channel activity to decline [110]. PIP2 likely binds to a pocket formed by the N- and C- termini of two adjacent Kir6.2 subunits [72, 111], directly interacting with positively charged residues in the slide helix near the lipid-cytoplasm interface to stabilize channel opening [109, 112–114]. The negatively charged head group and phospholipid tail of PIP2 are critical for channel activation by PIP2 because the PIP2 cleavage products-IP3 and diacylglycerol have limited effect [102].
Another class of anionic lipids, long chain fatty acyl-coenzyme A (LC-CoA) esters (e.g. oleoyl-CoA and palmityl-CoA), also potently activate KATP channels [115–121]. Long-chain acyl-CoA esters are β-oxidation products of fatty acids and the principal metabolic substrates of the heart which are thought to link KATP channel activity to the cellular metabolism of fatty acids. Through the same mechanism of phosphoinositides stimulation on KATP channels, LC-CoA esters increase KATP channel activity by decreasing ATP sensitivity and reducing channel rundown [117–120]. Acyl-CoA esters bind at Kir6.2 subunit with different binding sites from those for MgADP [116], but similar residues for PIP2 binding are probably involved [122] and an acyl-CoA ester binding motif reportedly locates in the C-terminus of Kir6.2 subunit [123].
Regulation by Other Metabolic Signals
Dynamically, KATP channel regulation is predominantly a balance between ATP-induced inhibition and MgADP-induced activation [12, 59, 124]. At saturating concentrations, MgADP shifts the IC 50 for ATP inhibition from ∼30 to ∼300 μM, which is still below the normal bulk cytosolic ATP concentration. KATP channels may respond to sub-sarcolemmal rather than bulk cytosol ATP concentration [125, 126]. Glycolysis has been reported to effectively inhibit channel activity, and functional glycolytic enzymes are associated with KATP channels [127], which suggests that glycolytic enzymes located in the membrane or adjacent cytoskeleton near the channels may be important in maintaining a high local cytosolic ATP/ADP ratio near KATP channels to inhibit channel activity [126]. In addition, phosphotransfer enzymes are recognized in amplifying small changes in bulk cytosolic ATP concentration, thereby regulating channel activity (Fig. 16.2). For example, the adenylate kinase (AK) phosphotransfer system facilitates conversion of ATP to ADP causing channels to open, whereas creatine kinase (CK) and protein kinase (PK)/glycolytic systems promote conversion of ADP to ATP and channel closure [61, 125, 128–130]. Competitive regulation between AK, CK, and PK regulates the conversion between ATP and ADP, which results in local changes of the ATP/ADP ratio in the vicinity of KATP channels, thereby regulating channel activity. Protein kinase A (PKA) [131, 132] and protein kinase C (PKC) [133] also increase channel activity and decrease ATP sensitivity through direct phosphorylation of KATP channels. Interestingly, AK [134], CK [135], lactate dehydrogenase (LDH) [135], glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [136] and PK [137] all physically interact with KATP channel subunits and this channel-enzyme complex may play an integral role in regulating the nucleotide concentration in the microenvironment surrounding the channel, and fine-tuning the response of KATP to the energetic state of the cell (Fig. 16.2).
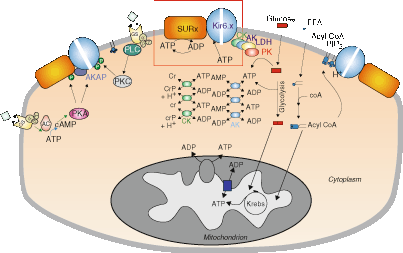
Figure 16–2.
The molecular regulation of cardiac KATP channels. The ratio of ATP to ADP levels is the major direct determinant of channel activity (box). Metabolic enzymes, including adenylate kinase (AK), creatine kinase (CK), and lactate dehydrogenase (LDH), in the cytoplasm and physically associated with the channel may serve to amplify metabolic changes, or locally buffer and control ATP/ADP levels, thereby fine-tuning channel activity. Non-nucleotide ligands, including PIP2, acyl-CoA, and H+, may also play a key role. PIP2 and acyl-CoAs have powerful stimulatory effects that are antagonistic to ATP inhibition. In addition, hormone receptor activation can lead to protein phosphorylation (P) with both stimulatory and inhibitory effects on the channel. However, none of these molecules acts in isolation, and the resultant KATP channel activity is an integrated response to a myriad of inter-related metabolic signals
The emerging picture of KATP regulation in the heart is complex. While ATP is a primary inhibitor, it is now clear that KATP activation is the result of dynamic regulation by multiple factors, which may account for the highly variable ATP sensitivity [62], the high variance of the latency to channel activation during anoxia [138], and the metabolic state-dependent inhibition by KATP channel blockers [139–141].
Cardiac KATP Channel Function
Cardiac KATP Channels and Cardioprotection
Severe metabolic stresses such as anoxia, metabolic inhibition and global ischemia cause marked channel opening, but under normal metabolic conditions, sarcolemmal KATP channels are predominantly closed, and therefore not expected to contribute significantly to cell excitability. However, due to the higher density of KATP compared to other sarcolemmal K+ channels, opening of as few as 1 % of KATP channels is expected to shorten cardiac action potential by about 50 % [142–145]. In so doing, KATP activation will reduce calcium entry and cause myocyte contraction to fail [146] and the energy stores that would otherwise be depleted in the contracting cell are preserved, providing cardioprotection.
In accordance with this cardioprotection concept, KATP channel openers enhance the preservation of ATP [147] and produce anti-ischemic effects by causing action potential shortening [148–150]. In parallel, glibenclamide prevents cardioprotective action potential shortening [151–153]. Renewed evidence for the cardioprotection by sarcolemmal KATP channels has been obtained through genetic manipulation. Moderate overexpression of SUR2A reportedly increases sarcolemmal KATP density and protects the heart against metabolic stress including hypoxia and ischemia/reperfusion [154]. Consistently, ischemic protection is abolished in rat cardiac myocytes transfected with a dominant-negative fragment of SUR2A which causes reduced sarcolemmal KATP expression [155]. Additionally, SUR1 overexpressing mice and Kir6.2−/− mice exhibit a disruption of KATP activity and impairment of the cardiac response to systolic overload following chronic transverse aortic constriction [156, 157].
However, in contrast to the accepted notion that increased KATP channel activity will produce cardioprotection, two recent studies have shown that neither SUR2−/− mice nor SUR1−/− mice have impaired response to ischemia reperfusion [158, 159]. SUR2−/− mice may actually be resistant to cardiovascular stress caused by global ischemia, due to reduced infarct size [158], contrary to the expected phenotype of impaired protection. However, these SUR2−/− mice were generated by disruption of NBD1, which might allow translation of the preceding TMD0 and TMD1 or following TMD2 of SUR2, and novel short form SUR2 proteins have been identified in both WT and SUR2−/− hearts [40]. The cardioprotection observed in these SUR2−/− mice may be due somehow to the presence of these short form proteins, but may also be related to abolition of the SUR2B component of vascular KATP channels [158]. Similar cardioprotection was also observed in SUR1−/− mice [159]. In an in vivo model of myocardial ischemia/reperfusion injury, SUR1−/− mice exhibit increased protection and reduced infarct size as well as preservation of left ventricular function [159], presenting the first potential pathological role for SUR1 in the heart. Again the mechanism is unknown, but may be related to abolition of SUR1, and hence abolition of KATP, in the atria, where SUR1 is an essential subunit [38, 48]. While it is not clear how SUR knockout mice produce enhanced cardioprotection, clearly reduction of either SUR1 or SUR2 expression can modulate cardioprotection, and hence these subunits may be useful cardioprotective targets.
Cardiac KATP Channels and Ischemic Preconditioning
Cardioprotective functions of the KATP channel have been best characterized during ischemic preconditioning. First recognized in 1986, this type of preconditioning arises from brief ischemic challenges that precede a prolonged insult, improving recovery of contractile function and reducing infarct size that results from more severe metabolic insult [160]. Among many endogenous ligands, such as adenosine and acetylcholine, and associated signaling proteins, such as protein kinase C and MAP or JUN kinases, KATP channels may be the common end-target in ischemic preconditioning [161]. Preconditioning can be mimicked by adenosine, as well as by synthetic KATP channel openers such as pinacidil and cromakalim [150]. Additionally, glibenclamide inhibits ischemic as well as adenosine- and acetylcholine-mediated preconditioning [150, 162]. Ischemic preconditioning is also abolished in hearts from Kir6.2−/− mice [156], in hearts from Kir6.2 gain-of-function mutation mice which show decreased KATP density [163], and in rat cardiac myocytes transfected with a dominant-negative fragment of SUR2A [155], strongly suggesting a role for sarcolemmal KATP channel as a preconditioning mediator.
In 1991, KATP channels were also reported in the mitochondria [164]. A succession of pharmacological studies that followed argued that mitochondrial rather than sarcolemmal KATP channels are primarily involved in preconditioning. First, action potential shortening cannot account for the cardioprotective effects of KATP channel openers [165–167]. Second, diazoxide, reportedly without effect on sarcolemmal KATP channels, can mimic ischemic preconditioning [168–170]. Third, 5-hydroxydecanoic acid (5-HD), reportedly a specific mitochondrial KATP channel blocker, effectively abolishes ischemic preconditioning and the anti-ischemic effects of KATP channel openers without inhibiting action potential shortening [171–173].
Several studies show that diazoxide activates sarcolemmal KATP channels [174, 175] and 5-HD suppresses APD shortening in ischemic hearts [152, 176], suggesting diazoxide and 5-HD actually target the sarcolemmal KATP channel. Moreover, additional KATP-independent targets are recognized as being sensitive to diazoxide and 5-HD: diazoxide inhibits succinate oxidation and succinate dehydrogenase activity; 5-HD serves as substrate for acyl-CoA synthetase [177]. Therefore, the use of the above pharmacological tools as probes for mitochondrial KATP requires reassessment.
Genetic manipulation has made it possible to circumvent some of the limitations of pharmacology by examining gene function directly. The molecular structure of the mitochondrial KATP channel is presently unknown, but Kir6.2, the primary pore-forming subunit of sarcolemmal KATP channels, does not appear to form mitochondrial KATP channels [178]. One study, using pharmacological tools and transfection systems suggests that current through the Kir6.1/SUR1 complex mimics the properties of mitochondrial KATP channels [50]. In this study, Kir6.1/SUR1 channels expressed heterologously in HEK293 cells were activated by diazoxide but not P-1075, and were inhibited by 5-HD but not HMR1098. The pharmacological properties of Kir6.1/SUR1 channels are similar to those reported for mitochondrial KATP channels [50], suggesting a Kir6.1/SUR1 composition in mitochondria. Therefore, genetically controlling Kir6.2 may be a direct means of assessing the role of KATP channels in the sarcolemma rather than in the mitochondria. Kir6.2−/− mice lose ischemia preconditioning [156] as well as diazoxide induced preconditioning [179]. Preconditioning is also abolished in cardiac Kir6.2 gain-of-function mice in which cardiac basal KATP activity is increased but overall activity is dramatically decreased [163, 180]. These studies are therefore more consistent with sarcolemmal rather than mitochondrial KATP channels mediating preconditioning. Ultimately, the existence of conventional KATP channels in mitochondria may be questionable: both Kir6.1 and SUR1 subunits may be expressed strongly at the sarcolemma of ventricular myocytes [41]. Even efforts to determine the molecular composition of mitochondrial KATP from immunohistological experiments may be unreliable given the still considerable difficulties in isolation of mitochondrial membrane proteins [181].
Cardiac KATP Channels and Arrhythmia
Lethal ventricular arrhythmia remains a leading cause of sudden death in patients with ischemic heart disease; 80 % result from ventricular fibrillation accompanied by myocardial ischemia and infarction [182]. The development of malignant arrhythmias during ischemia may be primarily due to the opening of KATP channels which cause rapid external potassium accumulation and reduction in action potential duration and therefore reduction in the refractory period in the ischemic myocardium [183, 184]. Additionally, dispersion of action potential duration may result from KATP activation: epicardial cells may be preferentially affected by KATP opening due to an action potential notch caused by the transient outward current (Ito) [185]. Depression of the membrane potential by Ito in conjunction with KATP channel opening will tend to prematurely deactivate Ca2+ channels, leading to early action potential termination. Selectively shortened action potentials in epicardium by KATP opening will then result in a transmural dispersion of repolarization, causing ST-segment elevation in the ECG and giving rise to phase 2 re-entry, which can precipitate ventricular tachycardia and ventricular fibrillation (VF) [185, 186]. In accord with this idea, Di Diego and Antzelevitch were able to show reentry in isolated ventricular myocardium after adding the KATP opener pinacidil, electrical homogeneity was restored and arrhythmias were abolished by blocking Ito (4-aminopyridine, 4-AP) or KATP (glibencamide) (Fig. 16.3) [186].
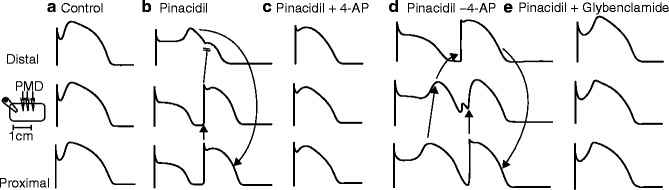
Figure 16–3.
KATP opening increases dispersion of repolarization. Action potential traces were recorded simultaneously from three sites along a canine epicardial preparation, P proximal, M middle, D distal. (a) Control traces recorded without any treatment. (b) Recorded after exposure to pinacidil shows the development of electrical inhomogeneity with loss of the dome at the two proximal sites but not at the distal site. The marked difference in repolarization times results in reentrant excitation at the proximal and middle sites. (c) Shows the effects of 4-aminopyridine (4-AP), a transient outward current blocker, in the continued presence of pinacidil. Inhibition of the transient outward current restores electrical homogeneity to the preparation, thus abolishing all arrhythmic activity. (d) Washout of 4-AP is attended by reappearance of electrical inhomogeneity and reentrant activity. (e) Illustrates the effects of glybenclamide, an inhibitor of the ATP-sensitive potassium current, in the continued presence of pinacidil. The effects of pinacidil were completely reversed, electrical homogeneity was restored, and all reentrant activity was abolished. The presumed circus movement pathways are indicated by the arrows (From Di Diego and Antzelevitch [186]. Reprinted with permission from Wolters Kluwer Health)
While KATP channel activation is potentially proarrhythmic and may predispose to lethal re-entrant arrhythmias, the opening of KATP channels during ischemia is also expected to stabilize the resting membrane potential and reduce ectopic pacemaker activity, i.e. an antiarrhythmic effect. Indeed, pharmacological studies provide two contrasting effects of KATP channels with respect to arrhythmias. Glibenclamide decreases the incidence of sustained tachycardia and ventricular fibrillation during ischemia-reperfusion [187–189], but increases the occurrence of tachycardia [190]. Similarly, some have observed that KATP channel openers promote tachycardia and fibrillation [58, 187, 190], while, others see a reduction in the overall incidence of arrhythmias [149, 191]. Grover et al. [149] reported opposite effects of the KATP opener cromakalim in different species: inhibition of fibrillation in anesthetized dogs but induction of fibrillation in isolated rat hearts, implying that the arrhythmic effects of KATP channels may be a function of the experimental model.
The significance of KATP channel composition in arrhythmia induction has been further explored through genetic manipulation. Kir6.2−/− mice exhibit high risk for induction of triggered activity and ventricular arrhythmia under catecholamine challenge, due to defective action potential shortening and predisposition to early afterdepolarizations [192]. Thus, sarcolemmal KATP is required for tolerance against triggered catecholamine-induced ventricular arrhythmia. Evidence for a proarrhythmic effect of sarcolemmal KATP channels comes from double transgenic (DTG) mice which overexpress both SUR1 and an ATP-insensitive Kir6.2 in the heart [193]. These SUR1-expressing DTG mice develop several different types of arrhythmias including AV block, atrial and ventricular tachyarrhythmia, indicating a proarrhythmic effect of KATP channels comprised of SUR1 and ATP-insensitive Kir6.2. In marked contrast, SUR2A-expressing DTG mice exhibit no change in arrhythmia incidence [193]. In addition, single transgenic mice which overexpress SUR1 but not SUR2A exhibit PR prolongation, suggesting delayed conduction in the AV nodal or diffuse conduction system. These results may reflect the higher sensitivity of SUR1-based KATP channels to metabolic activation [49], and indicate that expression of hyperactive KATP in the heart is likely to be proarrhythmic.
Also of interest are KATP mutations that confer risk for arrhythmia. The SUR2 T1547I mutation was associated with adrenergic atrial fibrillation originating from the vein of Marshall in a patient with paroxysmal atrial fibrillation [194]. More recent studies also report association of the S422L mutation in Kir6.1 with J-wave syndromes, which include Early Repolarization Syndrome and Brugada Syndrome (Fig. 16.4) [6–8]. As discussed above, the existence and distribution of Kir6.1 in the heart are not yet well established, nor can the consequences of mutations in other tissues be excluded, but these findings will certainly cause a re-evaluation of cardiac KATP channel function.
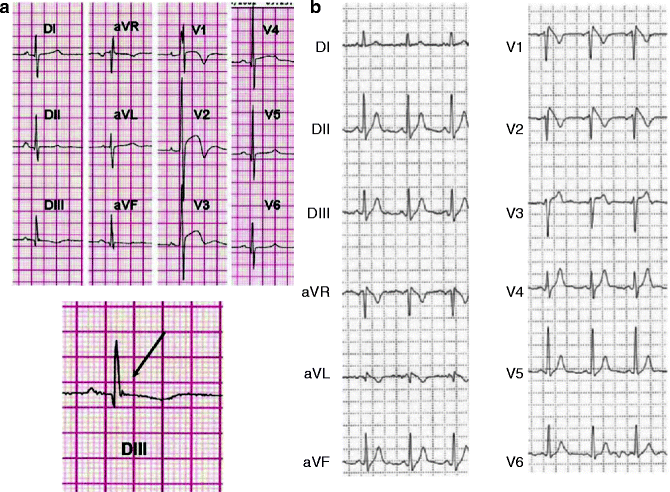
Figure 16–4.
ECGs in Kir6.1-associated J-wave syndrome. ECG recordings of one early repolarization syndrome case and one Brugada syndrome case associated with S422L mutation in Kir6.1 gene. (a) Twelve-lead ECGs of case 1 showing intraventricular conduction delay (V1), repolarization abnormalities in leads V1–V4 (left), and intermittent J waves in the inferior leads. Magnification of the J wave observed in lead DIII is shown at the bottom. (b) Spontaneous type 1 Brugada ECG pattern recorded during 24-h Holter monitoring in case 2 (From Medeiros-Domingo et al. [7]. Reprinted with permission from Elsevier Limited)
A role for sarcolemmal KATP channels during arrhythmias may be masked by the potential that mitochondrial KATP inhibition influences the incidence of arrhythmia. 5-HD suppressed the incidence of ventricular fibrillation induced by coronary ligation in rats [195], suggesting an antiarrhythmic effect for mitochondrial KATP inhibition. On the other hand, the antiarrhythmic effects of both Nicorandil and Minoxidil in rabbits were abolished by 5-HD but not by HMAR 1883, suggesting a proarrhythmic effect of mitochondrial KATP inhibition. The role of 5-HD in arrhythmias became more confusing when it was reported that 5-HD was not anti-arrhythmic in dogs [196] or in rats [197]. Conversely, HMR 1883 decreased the incidence of ischemia-induced irreversible ventricular fibrillation and improved survival during acute myocardial infarction in conscious rats as well as during reperfusion after brief myocardial ischemia in anesthetized rats [198], while in the same study, 5-HD did not protect hearts in these arrhythmia models [198], suggesting arrhythmias during ischemia and ischemia/reperfusion are the result of opening sarcolemmal KATP channels rather than mitochondrial KATP channels.
Cardiac KATP Channels and Cardiomyopathy
Cardiac hypertrophy can be an adaptive response but prolonged hypertrophy can progress into heart failure and sudden death, which may result from consequently disturbed electrical activity [199]. Decreased KATP channel responsiveness to ATP inhibition has been reported in hypertrophied cardiomyocytes suggesting hyper-activity [200, 201]. However, impaired KATP channel activation has also been reported in hypertrophic myocytes and suggested as a potential explanation for reduced tolerance of hypertrophied hearts to ischemia [202]. Interestingly, SUR2−/− mice exhibit greater ventricular mass and relative heart size compared to control cohorts, evidence of hypertrophy [158]. Kir6.2−/− mice also show increased mortality and exaggerated hypertrophy in response to pressure overload [157, 203] and to mineralocorticoid/salt challenges [204], suggesting that disrupting KATP channel actually predisposes hearts to hypertrophy under stress.
KATP channels appear to be less sensitive to ATP inhibition and more sensitive to KATP channel openers in failing human hearts, which may indicate that KATP remodeling may favor a beneficial effect in failing hearts [58, 205]. Optical mapping of failing hearts reveals increased APD shortening in response to diazoxide (Fig. 16.5). A heart failure model induced by transgenic expression of cytokine tumor necrosis factor alpha (TNFα) shows a disturbed link between the metabolic state of the cell and KATP channel activity and increased cardiomyocyte vulnerability to stress [206]. In this case, application of KATP channel openers improved stress tolerance [206]. The same group has reported linkage of human heart failure to KATP channel mutations. Mutations in SUR2 that cause decreased catalytic activity of NBD1 and disruption of KATP channel gating were identified in patients with dilated cardiomyopathy [9, 124]. Also, the Kir6.2 E23K polymorphism, which causes enhanced channel activity and is associated with type 2 diabetes susceptibility [207] also appears to be linked with subclinical maladaptive cardiac remodeling among individuals with increased stress load due to hypertension [208] and is over-represented in patients with congestive heart failure [5]. More recently, an in-frame deletion (E332del) and a missense mutation (V346I) were identified in two nonrelated sudden infant death cases. Both mutations lead to loss-of-function of Kir6.1 channels which may predispose a maladaptive cardiac response to systemic metabolic stressors [209].
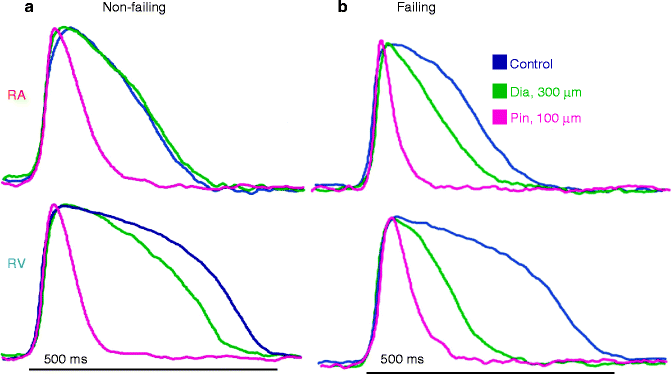
Figure 16–5.
Pathologic changes in human KATP channel opener responses. Optically mapped action potentials of human right atrial–ventricular preparation from a non-failing heart (a) and a congestive failing heart (b). A, In the non-failing heart, action potential in right atria (RA) is shortened by diazoxide but not pinacidil, while action potential in right ventricles (RV) is shortened by both diazoxide and pinacidil. (b) Shows diazoxide-induced action potential shortening in RA and a greater shortening in RV in the congestive failing heart, implying remodeling of KATP channel in failing hearts (From Fedorov et al. [58]. Reprinted with permission from Elsevier Limited)
Cardiac KATP Channels in Adaption to Physiological Stress
Potential protective effects of KATP are not limited to an extreme metabolic insult, such as global ischemia. A physiological role for cardiac KATP channels has been recognized during adaptation to physiological stress [1–3]. Short-term exercise up-regulates cardiac sarcolemmal KATP channel expression and enhances APD shortening which in turn reduces cardiac energy consumption in response to heart rate acceleration [1]. In addition to the lack of protection during ischemia-reperfusion, Kir6.2−/− mice exhibit intolerance to vigorous exercise in treadmill test and impaired survival in response to isoproterenol treatment compared with age- and gender-matched WT controls [3]. The Kir6.2−/− mice show less action potential shortening and diastolic calcium overload associated with contractile dysfunction which may explain the decreased adaption to stress [3]. Bulk adenine nucleotide content was not changed under the exercise and isoproterenol challenge, suggesting KATP channel activation may be caused by metabolic fluctuations in subsarcolemmal compartments. In addition, during chronic stress induced by long-term swimming training, Kir6.2−/− mice develop calcium-dependent structural damage and impaired contractile function, with a significant reduction in left ventricular fractional shortening and impaired cardiac output [2]. The Kir6.2−/− animals lack KATP in multiple tissues and further studies are warranted. However, selective disruption of KATP channels in heart (loss-of-function mutation of pore-forming subunit Kir6.1 [αMHC-Kir6.1AAA]) abolishes the enhanced APD shortening in response to the workload imposed by heart rate acceleration and causes increased cardiac energy consumption [1], strongly suggesting that KATP channels may play a more widespread role in cardiac physiology than generally appreciated, and are not limited to roles in pathological states.
Conclusions
Much has been learned about cardiac KATP channel since its discovery, but there are still significant gaps in our understanding. Recently, it has been recognized that cardiac KATP channel is chamber specific in mouse heart, but it is not clear whether this is universal for all species, whether there is any transmural heterogeneity or whether KATP channel composition differs in the conduction system. Many KATP channel mutations have now been identified as causal in human neonatal diabetes mellitus (NDM) and hyperinsulinism of infancy [210–212], but these patients have no reported cardiac abnormalities. Experimental and modeling studies have shown that activation of sarcolemmal KATP channels causes dramatic action potential shortening in vitro, but the function of cardiac KATP channels in vivo is not well established, even though there is emerging evidence that KATP channel mutations are linked to heart diseases. The debate regarding the role, and even the very existence, of mitochondrial KATP channels is still ongoing. Filling these knowledge gaps will require further study, and integration of results from basic cellular electrophysiology, to animal models and clinical disease.
Acknowledgement.
Our own experimental work has been supported by NIH grants HL45742, HL54171 and HL95010. We are grateful to the numerous former laboratory colleagues and collaborators for their contributions.
References
1.
Zingman LV, Zhu Z, Sierra A, Stepniak E, Burnett CM, Maksymov G, et al. Exercise-induced expression of cardiac ATP-sensitive potassium channels promotes action potential shortening and energy conservation. J Mol Cell Cardiol. 2011;51(1):72–81.PubMed
2.
Kane GC, Behfar A, Yamada S, Perez-Terzic C, O’Cochlain F, Reyes S, et al. ATP-sensitive K+ channel knockout compromises the metabolic benefit of exercise training, resulting in cardiac deficits. Diabetes. 2004;53 Suppl 3:S169–75.PubMed
3.
Zingman LV, Hodgson DM, Bast PH, Kane GC, Perez-Terzic C, Gumina RJ, et al. Kir6.2 is required for adaptation to stress. Proc Natl Acad Sci U S A. 2002;99(20):13278–83.PubMed
4.
Flagg TP, Enkvetchakul D, Koster JC, Nichols CG. Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev. 2010;90(3):799–829.PubMed
5.
Reyes S, Park S, Johnson BD, Terzic A, Olson TM. KATP channel Kir6.2 E23K variant overrepresented in human heart failure is associated with impaired exercise stress response. Hum Genet. 2009;126(6):779–89.PubMed
6.
Haissaguerre M, Chatel S, Sacher F, Weerasooriya R, Probst V, Loussouarn G, et al. Ventricular fibrillation with prominent early repolarization associated with a rare variant of KCNJ8/KATP channel. J Cardiovasc Electrophysiol. 2009;20(1):93–8.PubMed
7.
Medeiros-Domingo A, Tan BH, Crotti L, Tester DJ, Eckhardt L, Cuoretti A, et al. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes. Heart Rhythm. 2010;7(10):1466–71.PubMed
8.
Barajas-Martinez H, Hu D, Ferrer T, Onetti CG, Wu Y, Burashnikov E, et al. Molecular Genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8. Heart Rhythm. 2012;9(4):548–55.PubMed
9.
Kane GC, Liu XK, Yamada S, Olson TM, Terzic A. Cardiac KATP channels in health and disease. J Mol Cell Cardiol. 2005;38(6):937–43.PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


