Athlete’s heart
Hypertensive heart disease
Hypertrophic cardiomyopathy
Cardiac amyloidosis
Mitochondrial myopathy
Mucopolysaccharidosis
Anderson-Fabry disease
Onset age
Adolescence/adulthood
Adulthood
Adolescence/adulthood
Adulthood
Early childhood/adolescence
Early childhood/adolescence
Adulthood
Clinical presentation
No typical symptoms
Heart failure symptoms; typical angina; palpitations
No typical symptoms; dyspnea/angina at exercise in case of LVOT; syncope
Mostly symptoms of heart failure (despite normal LV function)
Systemic disease (no typical cardiac symptoms)
Systemic disease (no typical cardiac symptoms)
No typical symptoms
ECG
Possible high voltage QRS; resting bradycardia; ST segment abnormalities
Possible high voltage QRS; ST segment abnormalities
High voltage QRS; ST segment abnormalities
Absence of high ECG voltages despite LVH; conduction abnormalities
Dependent on subtype: conduction abnormalities; AV block
Conduction abnormalities; AV block
Conduction abnormalities; AV block
Echocardiography
Concentric LVH
Concentric LVH
Concentric assymmetric LVH
Concentric LVH; “sparkly” pattern; restrictive LV filling pattern; atrial enlargement; thickened cardiac valves and papillary muscles
Dependent on subtype: hypertrophic, dilative, restrictive pattern
Concentric LVH; valvular thickening with dysfunction (mostly aortic and mitral valve)
Concentric LVH; focal hypokinesia and wall thinning basal inferolateral; thickened aortic and mitral leaflets
CMR
Ischemic as well as nonischemic patterns of LGE
No specific LGE pattern; rather patchy pattern
Patchy or confluent LGE at the insertion points of the right ventricle into the LV and in the septum
LGE in the entire subendocardial circumference
Intramural LGE basal inferolateral in CPEO/KSS; unique HCM-like LGE in MELAS
No data
Subepicardial LGE basal inferolateral
EMB
Myocyte hypertrophy; expansion of interstitial and perivascular fibrosis by progressive collagen accumulation
Myocyte hypertrophy; expansion of interstitial and perivascular fibrosis by progressive collagen accumulation
Myocyte hypertrophy with nuclear enlargement; myofiber disarray and interstitial/replacement fibrosis; reduced arteriolar density
Green-appearing fibrils during Congo red staining under polarized microscopy; rodlike bundles in EM
Myocyte hypertrophy with vacuolated cardiomyocytes; proliferation of abnormal mitochondria; mosaic appearance of COX deficiency
Vacuolated myocytes with enlarged cytoplasm in LM; GAG laden cells (called “clear” cells)
Intramyocyte accumulation of glycosphingolipids; empty myocytes in LM; lamellar, dense bodies in EM
Extracardiac manifestations
None
Possible vascular complications
None
Nephrotic syndrome and renal insufficiency; cholestatic liver failure; malabsorption and gastrointestinal bleeding; peripheral or autonomic neuropathy; macroglossia or periorbital purpura
Dependent on subtype: ptosis, proximal myopathy, fatigue, exercise intolerance, encephalopathy, and ataxia
Growth retardation; skeletal deformities; CNS involvement; ocular and hearing impairment; respiratory difficulties; gastrointestinal disorders
Renal failure; corneal deposits; nervous, gastrointestinal, and cutaneous manifestations
Inheritance
–
–
Mostly autosomal dominant; rarely autosomal recessive, X linked or maternal
ATTR: autosomal recessive
Mostly maternal inheritance
Mostly autosomal recessive; only MPS II X linked
X linked: males diseased; females carriers
Specific treatment
–
Antihypertensive treatment strategies
–
Chemotherapy in AL
–
Stem cell transplantation; enzyme replacement therapy
Enzyme replacement therapy
Macroscopically, LVH is an increase of myocardial muscle mass. On a cellular level, there is considerable evidence that hypertrophy is caused by re-expression of many fetal genes and downregulation of adult genes. Re-expression of β-myosin heavy chain—most commonly found in areas of fibrosis and perivascular myocardial regions— and atrial natriuretic factor and α-skeletal actin is thought of as causally involved in the development of LVH. However, β-myosin heavy chain may simply be marker of LVH rather than a causative signal, because it was found in similar quantities both in hypertrophied and non-hypertrophied myocytes of hypertrophied rodent hearts [6]. Cellular myocyte hypertrophy involves both recruitment of contractile elements and myocyte proteins, but increases in myocardial mass also stem from an increase in cells that make up the connective tissue: fibroblasts, vascular smooth muscle cells, and endothelial cells. These changes of the extracellular matrix are key in the development of cardiac dysfunction and the clinical phenotype of the cardiomyopathy. There is a complex interplay between mechanical (hemodynamic) and neurohumoral stress inducing hypertrophic gene expression in hypertensive heart disease [7]. Abnormalities of the myocardial microvasculature cause an imbalance between oxygen delivery and the increased metabolic demands of the hypertrophied myocardium—functional ischemia causing anginal symptoms and perpetuation of cell death and myocardial fibrosis. In hypertensive heart disease, several molecular mechanisms and a few genetic determinants of a hypertrophic response have been identified [8]. In contrast, in familial HCM, several (but not all) genetic causes have been identified, and our understanding of the molecular mechanisms remains incomplete [9–11].
16.1.1.1 LVH in Hypertensive Heart Disease
Increased hemodynamic burden is a key stimulant for the development of LVH in hypertensive (and valvular) heart disease. LVH is an initially effective compensatory mechanism to overcome increased afterload to maintain constant wall stress [12]. While pressure overload invariably leads to concentric LVH, volume overload leads to eccentric hypertrophy with increases of left ventricular internal dimensions, which can also be seen in end-stage cardiomyopathy due to pressure overload [2, 13].
The anatomical classification proposed by Ganau et al. [14] is based on echocardiographic measurements of left ventricular geometry and left ventricular muscle mass. Left ventricular geometry is determined by relative wall thickness (RWT) calculated as doubling the width of the left ventricular inferolateral wall and divided by the left ventricular end-diastolic internal diameter in end diastole. A RWT ≥0.44 is diagnostic for concentric LVH, while a RWT <0.44 with increased left ventricular mass is indicative of eccentric remodeling. This category can be further distinguished from physiologic hypertrophy, which is characterized by mild increases of left ventricular mass and a RWT between 0.32 and 0.44 [12]. For the determination of left ventricular mass in this classification, the following formula is most commonly used:
![$$ \mathrm{Left}\kern0.24em \mathrm{ventricular}\kern0.24em \mathrm{mass}=0.8\times \left(1.04\times \left[{\left(\mathrm{LVIDd}+\mathrm{PWTd}+\mathrm{SWTd}\right)}^3-{\left(\mathrm{LVIDd}\right)}^3\right]\right)+0.6\kern0.24em \mathrm{g} $$](/wp-content/uploads/2016/07/A321847_1_En_16_Chapter_Equa.gif) Left ventricular mass is usually indexed to body surface area (Du Bois or Mosteller method) [15, 16]. LVIDd indicates left ventricular internal diameter in diastole, PWTd posterior wall thickness in diastole, and SWTd septal wall thickness in diastole.
Left ventricular mass is usually indexed to body surface area (Du Bois or Mosteller method) [15, 16]. LVIDd indicates left ventricular internal diameter in diastole, PWTd posterior wall thickness in diastole, and SWTd septal wall thickness in diastole.
In clinical practice, echocardiography is widely available, has a reasonable cost, and is accurate in the clinical setting for the determination of left ventricular mass. While magnetic resonance imaging has greater precision than echocardiography, it has a higher cost, more limited availability, and limited tolerability [17, 18]. Thus, echocardiography is still the primary method to assess the presence, magnitude, and hemodynamic complications associated with LVH.
The correlation between blood pressure measured in the physician’s office and left ventricular mass is not linear [19]. There are at least four explanations for this finding: (1) office blood pressure is not a reliable surrogate for overall hemodynamic burden, while 24-h ambulatory blood pressure is a much better surrogate and indeed correlates much closer with left ventricular mass [20]. (2) Both office and 24-h ambulatory blood pressure monitoring provide estimates of hemodynamic stress at one point in time, while the amount of lifetime hemodynamic stress clearly will determine the development of LVH to a much greater degree. Hypertensive heart disease is a chronic condition that develops over many years. (3) Neurohumoral stimulation linked to the development of LVH may differ between individuals with hypertension. (4) A genetic propensity for the development of LVH may exist both in hypertension and valvular disease. Racial/ethnic differences in the probability of developing LVH strongly suggest a genetic component [21–23].
16.1.1.2 Molecular Mechanisms of Hypertensive Heart Disease
(a)
Renin–angiotensin–aldosterone system (RAAS): Angiotensin II released by the myocardium activates G proteins and Rho proteins, which in turn increase protein synthesis in myocardial cells and collagen synthesis in fibroblasts [24–27]. These effects have been found to be independent of afterload in a mouse model suggesting a direct involvement of angiotensin II in LVH [28]. In addition, angiotensin II stimulates fibrosis via endothelin release [29]. Angiotensin II AT1 receptor blockers and angiotensin-converting enzyme (ACE) inhibitors effectively reduce LVH in hypertensive individuals corroborating the importance of the RAAS in the development of LVH [30] (Chap. 36). Aldosterone also seems to be involved in the development of hypertrophy. Mineralocorticoid receptors are abundantly expressed in cardiomyocytes [31], and aldosterone itself induces vascular [32] and myocardial inflammation [33], myocardial fibrosis [34], and LVH [35]. In a hypertensive model of endothelial dysfunction, eplerenone prevented cardiac inflammation and fibrosis [36]. The nonselective aldosterone antagonist spironolactone and the selective aldosterone receptor antagonist, eplerenone, provided clear clinical benefit in patients with systolic heart failure [37, 38], but the benefit is less clear in patients with diastolic heart failure, in whom LVH oftentimes was the common denominator [39, 40] (Chap. 38). Studies to better assess the clinical effectiveness of aldosterone blockers for the treatment of LVH are in process.
(b)
Endothelin–1: Endothelin-1, one of three human isoforms of endothelin, is a potent vasoconstrictor produced by endothelial cell. Endothelin has been shown to induce hypertrophy in animal models, and this phenotype can be suppressed by a pharmacologic endothelin-1 receptor blocker, such as bosentan [29, 41, 42]. Direct evidence of endothelin-1 as a mechanism for LVH in humans, however, is lacking (Chap. 45).
(c)
Heat shock proteins are intracellular proteins, which increase numerically in cells that are exposed to thermal or other forms of stress, and regulate nuclear transcription factors. These factors have been suppressed with gene therapy and antioxidant therapy producing an anti-hypertrophic effect even in the presence of pressure overload [43]. A proteasome inhibitor (PS-519) known to suppress heat shock proteins also prevented isoproterenol-induced LVH in animals with or without preexisting LVH [44].
(d)
G proteins: Many substances involved in the hypertrophic response to pressure and stress, including phenylephrine, angiotensin II, and endothelin-1, bind to myocyte membrane receptors that activate G proteins and small G proteins (i.e., Rho proteins). These proteins regulate transcription and have been shown to be involved in phenylephrine-induced LVH [45].
(e)
Calcineurin : It is a calcium-/calmodulin-dependent serine-threonine phosphatase that induces myocardial growth in response to different pathological stimulus. It dephosphorylates cytosolic factors (e.g., nuclear factor of activated T cell (NFAT)), enabling them to translocate to the nucleus to activate transcription. Transgenic mice that overexpress calcineurin or its transcription factor targets develop cardiac hypertrophy [46] (Chaps. 4 and 18).
16.1.2 Genetic Factors of Hypertensive Heart Disease
Heritability of left ventricular mass has been reported to be low in first-degree family members with the estimated heritability of adjusted left ventricular mass having values between 0.24 and 0.32 [47]. A markedly higher heritability of left ventricular mass of 0.59 was demonstrated in twins [22]. There can be a large variability of left ventricular mass in patients with similar office blood pressure. The high rates of LVH in certain race/ethnic populations [21] support a genetic predisposition for the development of LVH in response to pressure overload. There are now some genes that have been identified to correlate with LVH in hypertensive patients.
(a)
Corin, a membrane-bound serine protease expressed in cardiomyocytes, converts atrial and brain natriuretic peptide (ANP, BNP) to their active form. Corin knockout mice develop hypertension and cardiac hypertrophy [47]. Mutations of the corin I555 (P568) gene were exclusive to African-Americans in multiethnic samples with an allelic prevalence of 6–12 % and a clear association with both hypertension and LVH. Thus, corin mutations may explain in part the high prevalence of HTN and LVH in African-Americans [48].
(b)
(c)
Protein C overexpression causes progressive LVH and diastolic dysfunction in animals.
(d)
Bradykinin 2 receptor gene polymorphism, specifically the 9 bp receptor gene deletion is associated with greater left ventricular mass in subjects undergoing physical training [51]. Unlike LVH associated with pressure overload, the pathogenesis of LVH in HCM is clearly genetically mediated. However, genetic determinants of LVH in hypertensive hypertrophic cardiomyopathy may be important. As HCM can be associated with elevated LV systolic pressures due to left ventricular outflow tract (LVOT) obstruction, hemodynamics may also be a cause for exaggerated hypertrophy, and thus, there may be overlap between those two disease entities.
16.1.2.1 LVH in Hypertrophic Cardiomyopathy
Originally described in 1869 based on pathologic examination [52], HCM is present in 1 in 500 of the general population and the most common genetically transmitted cardiomyopathy. HCM can be caused by more than 1,400 different mutations [53] and is usually transmitted in an autosomal dominant pattern. Familial HCM describe a phenotype of thickened myocardium (≥15 mm) in the absence of increased afterload (such as hypertension or aortic stenosis) or other explanations for the thickened myocardium (see Table 16.1). A genetic abnormality in gene loci encoding sarcomere proteins is often present (Table 16.2), and over 1,400 gene variations have been linked to HCM. However, in more than 50 % of probands tested, a causative gene cannot be identified [54], underscoring the complexity and variability of the genetics in HCM. Figure 16.1 illustrates the structure of proteins involved in HCM-causing mutations. Mutations in two genes—MYH7 and MYBPC3—account for as many as 75 % of HCM gene-positive individuals [11]. A small subgroup of gene-positive HCM patients has two or more sarcomere protein gene mutations, which may be associated with an earlier onset and/or more rapid disease progression [55, 56]. The utilization of genetic testing is increasing. We believe the most useful applications of genetic testing are twofold: (1) in patients with a clinical suspicion of HCM, the disease can be confirmed, and (2) in family members of an affected gene-positive HCM patient, the presence of the gene defect can be confirmed or excluded. The latter situation provides assurance to gene-negative family members preventing unnecessary serial testing of these individuals, reducing stress, anxiety, and health-care costs [54]. In contrast, patients who are diagnosed with HCM in childhood or adolescence will need to be followed closely for the development of LVH. Whatever the genetic constellation, the clinical phenotype is highly variable, even within families of the same gene mutation, ranging from an asymptomatic course without macroscopic evidence for disease to an individual who has severe and rapid progressive LVH and cardiomyopathy and possibly early sudden cardiac death [54]. Figure 16.2 shows necropsy specimens from the hearts of two patients with HCM: one who had asymmetric septal hypertrophy and one who had concentric LVH. Some studies have identified high-risk [57, 58] and low-risk [59] mutations; however, conflicting reports exist [60] regarding the prognostic capability for predicting SCD. At present there is a lack of consensus about the role of genetic testing in predicting the magnitude or progression of LVH or the development of LVOT gradients, mitral regurgitation, or the clinical course and status as well as the need for strategies to prevent sudden death such as an implantable cardiac defibrillator (ICDs) [54, 61]. The relatively high cost of genetic testing, the inability to detect a disease-causing mutation in phenotypically affected patients (up to 50 %), and genetic variations of uncertain significance remain a challenge when using genetic testing in clinical practice [54].
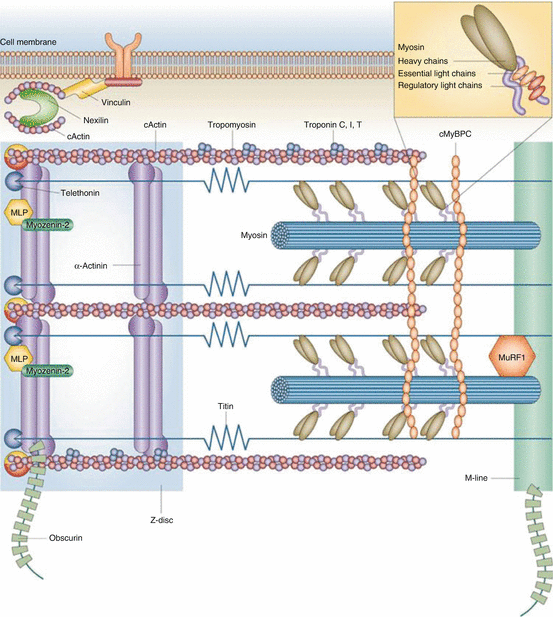
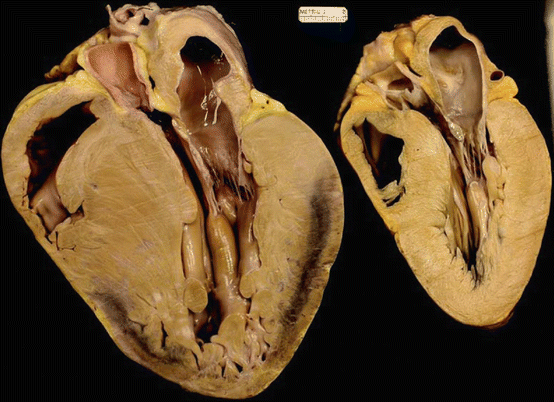
Table 16.2
HCM-associated genes
HCM gene | Protein defect | Estimated prevalence in HCM probands (%) | Strength of evidence for causality | Location |
|---|---|---|---|---|
No gene identified | NA | 50 | NA | NA |
MYBPC3 | Cardiac myosin-binding protein C | 15–25 | ++ | Intermediate filament |
MYH7 | β-Myosin heavy chain | 15–25 | ++ | Thick filament |
TNNT2 | Cardiac troponin T | 7 | ++ | Thin filament |
TNNI3 | Cardiac troponin I | <5 | ++ | Thin filament |
TPM1 | α-Tropomyosin | <5 | ++ | Thin filament |
MYL3 | Myosin light chain 3 | <1 | ++ | Thick filament |
MYL2 | Cardiac regulatory myosin light chain | <2 | ++ | Thick filament |
MYH6 | α-Myosin heavy chain | <1 | + | Thick filament |
TNNC1 | Cardiac troponin C | <1 | ++ | Thin filament |
ACTC | a-Actin | <1 | ++ | Thin filament |
MYOZ2 | Myozenin-2 | <1 | + | Z-disc |
ACTN2 | Alpha-actinin-2 | <1 | + | Z-disc |
CSRP3 | Cysteine and glycine-rich protein 3 | <1 | + | Z-disc |
TCAP | Telethonin | <1 | + | Z-disc |
CASQ2 | Calsequestrin | <1 | + | Ca++ handling |
JPH2 | Junctophilin 2 | <1 | + | Ca++ handling |
Fig. 16.1
Structure of proteins involved in HCM-causing mutations. cActin α-cardiac muscle actin 1, cMyBPC cardiac myosin-binding protein C, MLP cysteine and glycine-rich protein 3 (muscle LIM protein), MuRF1 E3 ubiquitin-protein ligase TRIM63 (muscle-specific RING finger protein 1) (Adapted with permission from Frey et al. [11])
Fig. 16.2
Two heart necropsy specimens from teenagers with hypertrophic cardiomyopathy who had sudden death. Both specimens demonstrate left ventricular hypertrophy; left, asymmetric septal hypertrophy in the specimen; right, concentric hypertrophy in the specimen (Adapted with permission from Roberts [154])
As seen in Fig. 16.3, HCM is associated with cardiomyocyte hypertrophy, myofiber and myofibrillar disarray with more or less pronounced interstitial fibrosis, and an abnormal microvasculature with intimal hyperplasia and medial thickening are characteristic of HCM. The magnitude of myofiber disarray in HCM is quantitatively increased in comparison with other conditions with LVH [62]. Animal models including the use of transgenic mice [63, 64], transgenic rabbit [65], Maine coon cats [66], and zebrafish [67] are being used to study the molecular mechanisms that translate a genetic defect into the hypertrophic phenotype. Several of these mechanisms have been proposed as being responsible for the clinical phenotype of HCM patients, and it is thought to be likely that HCM is a multifactorial disease:
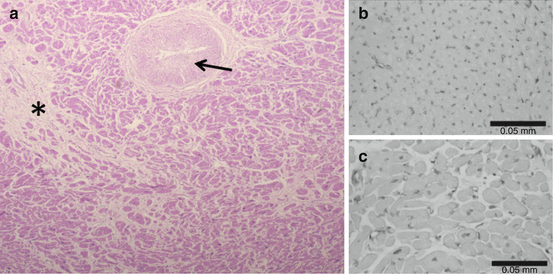
Fig. 16.3
Myocyte disarray, interstitial fibrosis (*), and vascular remodeling with intimal hyperplasia and medial thickening are typical for HCM but also other forms of LVH (panel a). Myocardial capillary density (panel c) is markedly decreased in HCM compared to a normal heart (panel b) (Sources: Panel a: Robert J Siegel, MD. Panels b and c: Adapted with permission from Kofflard et al. [155])
(a)
Depressed contractile function: One mechanistic hypothesis attributes myocardial contractile dysfunction from myocyte disarray and changes of the connective tissue (i.e., fibrosis) as the cause for progressive hypertrophy in the form of a compensatory mechanism [68–72]. There are several factors that cannot be explained by this compensatory hypothesis: (1) Hypertrophy often develops or progresses during the physiologic growth phase with and after puberty with no or slow progression thereafter even if diastolic and sometimes systolic function worsens. The protein defect on the other hand is present since heart development; thus, a purely compensatory mechanism seems unlikely. (2) Hypertrophy usually is asymmetric (but not always), while compensatory LVH is generally concentric (but not always). (3) A gain in function of the affected myocardium has been observed more recently with an increased energetic cost of cardiac contraction rather than left ventricular dysfunction as discussed next [73].
(b)
Abnormal calcium handling: Several animal studies suggested that hypertrophic myocytes exhibit an increased sensitivity and affinity to Ca2+ of the mutated proteins causing increased cross-bridge turnover and actin-activated ATPase activity [73–75]. This gain of function creates a greater energetic cost of each contraction, and energy depletion subsequently leads to cell death [73]. The enhanced calcium sensitivity has also been shown to increase susceptibility to ventricular arrhythmia by shortening the effective refractory period, increasing the heterogeneities in ventricular conduction and delayed after depolarization [74, 76] (Chap. 4).
(c)
Myocardial fibrosis: Myocardial fibrosis appears to be the result of premature cell death and expansion of interstitial cells and proteins. Premature myocyte demise is thought to be related to changes in the microvascular architecture in conjunction with the aforementioned abnormal energy homeostasis. One key signal for fibroblast stimulation is the transforming growth factor-β (TGF-β). Indeed suppression of TGF-β [64] with the angiotensin II AT1 receptor antagonist losartan [77] decreased fibrosis in animal models. The Valsartan for Attenuating Disease Evolution in Early Sarcomeric HCM [VANISH] trial [78] is testing whether angiotensin II AT1 receptor blockers can reduce myocardial fibrosis in HCM patients. Late gadolinium enhancement on cardiac magnetic resonance imaging (MRI) identifies the extent of myocardial fibrosis and scarring and is associated with an increased risk of sudden death [54, 79]. Serum by-products of fibroblast-secreted collagen are elevated in HCM patients with hypertrophy, in gene-positive probands without hypertrophy and in HCM patients without MRI evidence of scarring. These findings suggest that myocardial fibrosis could be an early causative pathophysiologic mechanism in the development of hypertrophy, rather than a result of hypertrophy [80].
(d)
Abnormal biomechanical sensing: Some of the HCM mutations involve proteins linked to the Z-disc and the M-band as shown in Table 16.2. These regions have been described to exert hypertrophic signaling from stretch-sensitive transcriptional modifiers in response to biomechanical stress [81]. Alterations in these regions may be involved in stimulating local hypertrophy. This hypothesis, however, is largely speculative at this time [11].
(e)
Abnormal energy production: Several observations suggest an abnormal supply and demand of cardiac sarcomere adenosine triphosphate [ATP] which is central to contraction and relaxation (myosin ATP) and calcium homeostasis (sarcoplasmic reticulum). 31Phosphorus-NMR demonstrates abnormal cardiac energetics in both HCM patients and gene-positive probands when compared to healthy controls; this finding suggests that altered ATP production and use could be a causative factor in the development of the HCM phenotype [75]. Furthermore, mitochondrial abnormalities can be observed in gene-positive individuals without a hypertrophic phenotype, which suggests that these altered energetics in HCM and are a cause, but not the result, of hypertrophy [82]. As discussed in Table 16.1, the same mechanism is found to cause myocardial hypertrophy in genetic diseases, which also causes mitochondrial dysfunction. Abnormal cardiac energetics cause diastolic dysfunction from deficient calcium reuptake in the sarcoplasmic reticulum and a state of calcium overload in systolic and diastolic heart failure [83]. These findings are consistent with the clinical presentation of many HCM patients [73].
(f)
Abnormal microvasculature: Scar burden (by cardiac MRI) and microvascular dysfunction appear to be closely related in HCM patients, and they have more pronounced gene-positive than gene-negative probands [84]. Histologically, intramyocardial microvessels demonstrate a completely abnormal architecture. Figure 16.3 shows histological findings of intimal hyperplasia (“onion skin appearance”), media hypertrophy, and decreased capillary density, all of which leads to reduced microvascular blood flow. In this combination with increased metabolic demands of the hypertrophied myocardium this causes microvascular ischemia and cell death. Therefore, this microvascular ischemia perpetuates fibrosis and as a consequence compensatory hypertrophy and contributes to the pro-arrhythmic substrate. In addition, the presence of microvascular ischemia indicates a worse prognosis in HCM [85]. Ongoing clinical trials are investigating improvements in microvascular ischemia and related arrhythmias from the use of late-current sodium channel blockers [86, 87].
16.1.2.2 Diagnosis of Hypertrophic Cardiomyopathy
Echocardiography is the standard method to screen for HCM because it has a good sensitivity for detecting global or focal hypertrophy. Therefore it is also the test of choice to follow affected individuals, who carry a diagnosis of HCM as well as screening their family members who are at risk for the development of HCM. Furthermore, Doppler echocardiography provides essential hemodynamic data identifying abnormal LV filling, LVOT gradients at rest and during exercise, and mitral regurgitation, which is associated with LVOT gradients and systolic anterior motion of the mitral valve (SAM). Echocardiography is also an essential method to monitor the mechanisms of symptoms, the risk of sudden death, as well as the effects of treatment, and the changes in resting and exertional LVOT gradients, mitral regurgitation, diastolic function, and pulmonary artery systolic pressure. Because of its better resolution and lower inter-study-variability than echocardiography [17, 18], MRI is increasingly being used to evaluate HCM patients; in addition, MRI has better spatial resolution, is less operator dependent, and is not affected by chest wall configuration or body habitus. Furthermore, late gadolinium enhancement (LGE) can detect the presence, the pattern of distribution, and the extent of myocardial fibrosis, which is a marker for potential ventricular arrhythmias [88]. However, the importance of quantification of cardiac fibrosis as an indication for primary prevention of sudden death with implantable defibrillators is controversial and focus of ongoing research. While MRI has greater sensitivity to detect abnormal wall thickness and provide precise quantification, its high cost and poor tolerability in some patients limit its use for longitudinal follow-up. In addition, echocardiography is superior to MRI for the assessment of mitral regurgitation, quantification of LV outflow tracts, pulmonary artery systolic pressure, and left ventricular filling pressures.
Endomyocardial biopsy with histologic evaluation of myocyte appearance and myofiber orientation is useful for excluding other infiltrative cardiomyopathies which can mimic HCM as detailed in Table 16.1. As previously discussed, genetic testing is helpful to confirm clinical suspicion of HCM and to exclude a known gene defect in family members of an affected individual, which obviates the need for serial long-term follow-up. At present, genetic testing is not a reliable method to predict future development of the disease in phenotypically normal but gene-positive individuals or for the prediction of disease progression and severity in HCM patients [54].
16.1.2.3 Amyloidosis as a Cause of Cardiac Hypertrophy
Cardiac amyloidosis causes a progressive increase in heart wall thickness that is not due to myocardial hypertrophy but rather to extracellular amyloid deposition [89]. This extracellular deposition of amyloid fibrils is composed of an autologous protein which has a beta sheet fibrillar confirmation. While the two main forms of amyloidosis, light chain (AL) and TTR amyloidosis, are the most common, there are more than 30 related amyloidosis proteins capable of forming amyloid fibrils [90, 91]. Cardiac involvement in amyloidosis can be rapidly progressive, and it is the most common cause of death in AL amyloidosis and a major determinant of prognosis. AL amyloid frequently involves the heart, liver, kidney, peripheral and autonomic nervous system, as well as the GI tract [89, 90]. Most patients are diagnosed in the fifth decade of life, and about 50 % have cardiac involvement. CHF augurs a poor prognosis with a survival of only 6 months in untreated patients. Death is usually due to progressive CHF or sudden death due to asystole or electromechanical dissociation.
For TTR amyloidosis, there is a hereditary and nonhereditary form, the latter of which is known as senile amyloid (SA). The hereditary form is autosomal dominant with a 50 % likelihood that the offspring will inherit the disease. TTR generally manifests in the third to fifth decades as cardiac amyloid, neuropathy, or both, depending on the specific molecular abnormality. Senile amyloidosis is related to the breakdown of abnormal TTR. SA generally affects the heart in men in their seventh or eighth decade of life [92]. CHF is often the first manifestation of SA.
Amyloidosis has protean manifestations due to organ infiltration, and symptoms are often nonspecific. Amyloid may present as CHF, progressive wasting, or as a peripheral neuropathy. The finding of low voltage in the electrocardiogram and thick LV walls in the echocardiogram known as voltage-mass discordance is a useful clue to the diagnosis of amyloid in a CHF patient [89]. Cardiac MRI with abnormal gadolinium uptake may also be indicative of cardiac amyloid [93]. Technetium pyrophosphate radionuclide scans may show homogenous uptake of the heart in cases of TTR and SA due to the binding of the P component of the amyloid fibrils [89]. Once there is clinical suspicion of amyloid, a tissue diagnosis should be made. This can be done with a needle biopsy of the abdominal fat or biopsy of another involved tissue. However, in cases with suspected cardiac amyloid, an endomyocardial biopsy may be needed [93]. For AL, amyloid blood and urine are also assessed with immunofixation to detect abnormal proteins which can be quantified by a free light chain assay. In the presence of these abnormal proteins, a bone marrow biopsy should be done to assess the severity of plasma cell dyscrasia [90]. In the absence of AL amyloid, blood testing can be done for a mutation of TTR. If this is negative, SA is the most likely diagnosis. In ambiguous cases, special staining of the biopsy specimens can elucidate the type of amyloid.
Cardiac amyloidosis can be clinically distinguished from HCM by the progressive nature of LVH secondary to progressive amyloid deposition. In HCM the ECG demonstrates LVH, whereas in amyloid, the ECG voltage is low and progressively decreases with amyloid infiltration as wall thickness increases due to the amyloid infiltration [93]. The clinical presentation of cardiac amyloid reflects myocardial infiltration. Initially there is impaired diastolic dysfunction and which generally progresses to systolic dysfunction [89]. Patients often develop right- and left-sided heart failure as well as atrial and ventricular arrhythmias [94, 95]. When CHF is seen in association with other organ involvement suggesting amyloid infiltration such as macroglossia, carpal tunnel syndrome, easy bruising and bleeding, autonomic neuropathy, nephrotic syndrome, and cachexia, amyloid should strongly be considered.
On echocardiography, amyloid may mimic HCM by the presence of asymmetric septal hypertrophy. Cardinal echocardiographic findings in amyloid to suggest the diagnosis are a sparkling appearance of the myocardium, progressive diastolic dysfunction, increased wall thickness of the LV, RV septum in the absence of systemic or pulmonary hypertension, and the intra-atrial septum and bi-atrial dilation. Strain and strain rate imaging show impaired myocardial function which progresses over time [93].
Treatment which is similar for all amyloidosis involving the heart requires management of the cardiac-related complications. Most patients develop CHF with volume overload and thus should be on a low-sodium diet (1–2 G of sodium per day). Patients should monitor their weights daily as well as their edema and ascites. Diuretics are the mainstay of therapy for CHF due to amyloid [89]. In addition certain cardiac drugs such as digoxin and calcium channel blockers are generally contraindicated as they might bind to the P component of the amyloid fibrils which can result in digoxin toxicity and in the case of calcium channel blockers severe and even fatal hypotension. Beta blockers are generally not useful in amyloid as they promote bradycardia as well as hypotension. Afterload reduction is often problematic due to autonomic dysfunction and systemic or orthostatic hypotension [89]. Chronic oral anticoagulation is warranted in patients with atrial fibrillation to reduce the risk of systemic embolization and stroke. TTR has a proclivity for cardiac conduction disturbances and bradycardia so that pacemaker therapy is useful in this setting. It is unclear if defibrillators are effective in preventing sudden death in these patients as sudden death may be due to asystole or electromechanical dissociation [94, 95].
The most common type of AL amyloid produced by plasma cells in the bone marrow may be treated by autologous bone marrow transplantation or chemotherapy with thalidomide, melphalan, dexamethasone, bortezomib, lenalidomide, and bendamustine or with a combination of these medications [90]. AL amyloid therapy is focused on reducing or eliminating plasma cell production of free light chains. Response to therapy can be in part assessed by reduction in the cardiac biomarker for CHF (BNP). A 30 % reduction in levels is indicative of a positive response; however, progressive renal failure can cause increases in BNP and limit the utility of the BNP level for assessing the response to therapy [91].
Melphalan-dexamethasone is a standard regimen which is generally well-tolerated [96], but its effectiveness in patients with advanced cardiac disease is limited [91], with median survivals being between 10 and 18 months. Recently, bendamustine, an alkylating agent with a unique mechanism of action, has shown potential promise as a therapeutic agent. However, the data is limited on improving survival.
Combination therapy with immunomodulating drug thalidomide with dexamethasone and cyclophosphamide has shown benefits on end-organ responsiveness in 33 % of patients; however, toxicity occurs in up to 60 % of patients. The second-generation drug lenalidomide and third-generation agent pomalidomide in combination with dexamethasone are being assessed [90]. In small studies, 40–50 % of patients appear to be responders. The addition of alkylating agents increases the response rate but also the drug toxicity [91].
Protease inhibitor drugs such as bortezomib, ixazomib, and carfilzomib are being tested [97]. These drugs have been shown to be effective in reducing the production of free fibrillar light chains [98]. The newer protease inhibitors ixazomib and carfilzomib appear to have a greater protease inhibitor effect and less toxicity. These drugs are currently being evaluated in clinical trials [97].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


