Fig. 1.1
Cardiac cytoarchitecture changes during development and in disease. Schematic representations of an embryonic cardiomyocyte (a) showing its polygonal shape, an adult cardiomyocyte (b), which is rod-shaped and a cardiomyocyte from a DCM heart (c), which is characterised by a more irregular shape. The myofibrils are shown as black striations; the nuclei are shown in blue. While the embryonic cardiomyocytes make direct cell–cell contact all around their surface (turquoise signal), these become restricted to the bipolar ends of mature cardiomyocytes postnatally. DCM is characterised by an increased membrane convolution and elevated expression of actin-anchoring proteins, which is depicted as a thickening of the turquoise line
How are the different cytoskeletal multiprotein complexes assembled in the heart? As mentioned above, cross-striated myofibrils characterise the first beating cardiomyocytes. The basic unit of a myofibril is a sarcomere, which is defined as the region between two neighbouring Z-discs (Fig. 1.2). The actin-containing thin filaments are anchored at the Z-disc, mainly via a protein called alpha-actinin (Ribeiro Ede et al. 2014). The thick filaments, which consist of myosin and associated proteins, are found in the middle of the sarcomere, where they are anchored to the elastic filament system composed of titin in a structure called the M-band. During myofibrillogenesis the first organised complexes that can be detected consist of concentrations of sarcomeric alpha-actinin and the N-terminus of titin, which are organised along filamentous actin with a regular distance from each other in close proximity to the plasma membrane (Sparrow and Schöck 2009). Study of the process of myofibrillogenesis in cultured cardiomyocytes has identified several precursor structures such as IZI brushes or premyofibrils (for review see Sanger et al. 2005). However, non-muscle myosin, which defines a premyofibril (Rhee et al. 1994), is not seen in between the alpha-actinin–titin complexes in the embryonic heart in situ and also the distance between them is wider than the distance observed in premyofibrils or stress fibre-like structures (Ehler et al. 1999). This is in contrast to myofibrillogenesis in skeletal muscle of the embryo, where two distinct populations of assembling myofibrils can clearly be distinguished in zebrafish somites (Sanger et al. 2009). Also sarcomeric protein expression during heart development is not switched on sequentially like it is observed for skeletal muscle (Ehler et al. 1999; Fürst et al. 1989); therefore the process of differentiation and thus myofibrillogenesis in the embryonic heart in situ may be too rapid to allow or require precursor structures (Ehler and Gautel 2008).
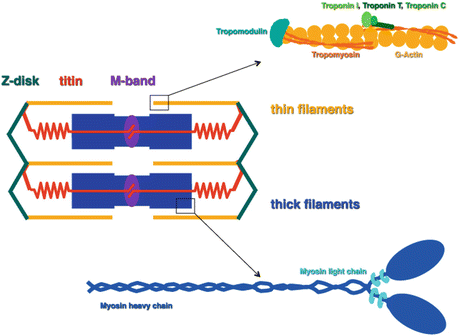
Fig. 1.2
Simplified schematic representation of a sarcomere, the basic unit of a myofibril. A sarcomere is defined as the region between two Z-discs, where the thin (actin) filaments are anchored. The M-band is a structure in the very middle of the sarcomere and serves for the integration of the titin filaments with the bipolar myosin (thick) filaments. An enlarged scheme of the pointed end of a thin filament is shown in the top right, depicting the actin subunits and their associated proteins tropomyosin and the troponins. The bottom right shows a myosin molecule, which is composed of two myosin heavy chains and four myosin light chains. Myosin binding protein-C is associated with a subset of the myosin molecules but was omitted from this scheme for reasons of clarity
The assembly of the specialised type of cell–cell contact, the intercalated discs, is only achieved postnatally in the heart. The intercalated discs are classically defined as having three types of contact (Forbes and Sperelakis 1985): adherens junctions that anchor actin filaments (or in the case of muscle cells the myofibrils), desmosomes that anchor the intermediate filaments (desmin) and gap junctions, which are composed of connexin-43 channels and mediate intercellular communication (Fig. 1.3). Embryonic cardiomyocytes have all three types of cell–cell contact distributed all around their plasma membrane and restriction of contact sites to the intercalated disc proper only happens around birth (Hirschy et al. 2006). Establishment of mature gap junctions happens even later than that (Angst et al. 1997). Recently the concept of a separate existence of adherens junctions and desmosomes at the intercalated disc has been challenged and a novel type of contact was proposed, the area composita. This is based on the observation that in the heart there is no strict segregation between the distribution of classical adherens junction and desmosomal proteins and that a mixed composition of these junctions is predominant (Franke et al. 2006).
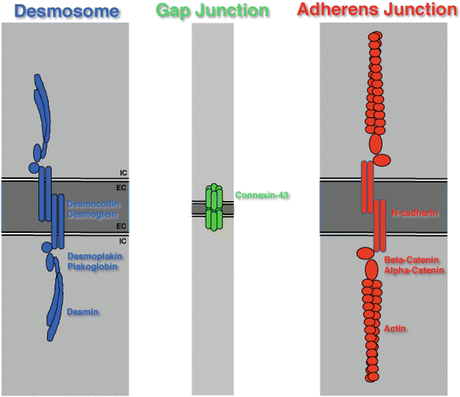
Fig. 1.3
Simplified schematic representation of the different contact structures at the intercalated disc. Desmosomes, gap junctions and adherens junctions are depicted. Desmosomes link up to the intermediate filament cytoskeleton, which is composed of desmin in muscle cells, while adherens junctions link up to actin filaments (myofibrils). This scheme shows a minimalist version of these structures and many associated proteins were omitted for reasons of clarity. IC intracellular, EC extracellular
1.2 Cardiomyocytes in Disease
Apart from loss of cardiac tissue as for example by ischemia as caused by a myocardial infarction, two major forms of cardiomyopathy can be defined, hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM). Another type of cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy (ARVC), is less frequent, but also associated with changes in cytoarchitecture (Basso et al. 2012). As we accumulate more data from next generation sequencing, the frequent hereditary component of these diseases becomes more evident and is no longer restricted to huge families. On a macroscopic basis HCM is defined as a thickening of the ventricular and/or septal wall and thus a reduction in chamber size and blood volume at filling (Seidman and Seidman 2001). DCM is characterised by a ballooning heart with an increased diameter of the ventricle (Seidman and Seidman 2001). As sudden cardiac death, especially in young athletes, can be the first sign of a HCM or an ARVC, more knowledge on the genetic cause of these diseases and screening for known variations can be potentially life-saving (Basso et al. 2012). Classically HCM is defined as being caused by mutation in proteins that constitute the myofibrils, while DCM is often associated by mutated cytoskeletal proteins (Chien 1999). The most frequent mutations that cause HCM are seen in sarcomeric myosin heavy chain and truncations of myosin binding protein-C (Frey et al. 2012), while truncating mutations in titin were initially seen in DCM (Gerull et al. 2002) and recently proposed to be among the main causes for DCM (Herman et al. 2012). However, the more sequence data are accumulated the more obvious it becomes that these simple associations are not correct (for recent review of disease causing mutations see McNally et al. 2013) and that for example proteins such as MLP and AMPK, which are not integral myofibrillar proteins, can be mutated in HCM, while mutations in classical myofibrillar proteins such as myosin can also lead to DCM. Truncating mutations in titin were also found to be associated with all types of cardiomyopathies (McNally et al. 2013). A more clear-cut link seems to exist at present for ARVC, which has been named the disease of the desmosome, a special type of cell–cell contact that links the intermediate filament cytoskeleton to the plasma membrane and is seen as mainly playing a structural role (Rickelt and Pieperhoff 2012). However, also for ARVC the situation will become more complex the more next generation sequencing (NGS) data are available and already now a link between the developing disease and Wnt signalling, which is usually active mainly in early embryonic heart development, is proposed (Asimaki et al. 2009; Swope et al. 2013).
At the level of the cardiomyocyte, HCM and DCM present quite different phenotypes (Fig. 1.4). HCM is usually already evident by Hematoxylin and Eosin staining and presents as myofibrillar disarray (Frey et al. 2012). The cardiomyocytes have a larger diameter and there is also a change in the arrangement of the tissue, with cell–cell contacts as characterised by the adhesion junction protein beta-catenin also being found at the sides of the cells. Apart from obvious regions of necrosis, DCM tissue can appear relatively unharmed at the histological and ultrastructural level and it required the careful analysis of the first mouse model for this disease, the MLP knockout mouse (Arber et al. 1997), to get a grip on the alterations that happen at the cellular level in this disease (Ehler et al. 2001). Freshly isolated cardiomyocytes from MLP knockout hearts are characterised by a more irregular shape compared to healthy hearts (see Fig. 1.1c), but there is no consistent cellular elongation, which might explain the ballooning ventricles but rather a general loss of size control (Leu et al. 2001). Similar observations were also made in other mouse models for DCM and even in samples from human DCM patients, suggesting that this may be a more general phenomenon of the disease (Hirschy et al. 2010; Pluess et al. 2015). With the obvious consequences that this has on the three-dimensional arrangement of heart tissue, it may provide some of the explanations for the alterations that are seen in the ways the cells make contact with their extracellular matrix. For example, the costameres become disorganised in the MLP knockout mouse and also the way the extracellular matrix is distributed over the cardiomyocytes changes in cardiomyopathy (Ehler 2010; Ehler et al. 2001; Hoskins et al. 2010). This probably reflects a loss in membrane organisation especially as far as triads are concerned and was also reported by ion conductance microscopy on chronic heart failure samples (Nikolaev et al. 2010). While the organisation and composition of the myofibrils is much less affected in DCM compared to HCM and there is for example no consistent re-expression of embryonic isoforms of the contractile proteins actin and myosin (MacLellan and Schneider 2000), there is a shift towards the expression of more compliant isoforms of elastic proteins that are needed to maintain a working sarcomere such as longer versions of titin and the EH-myomesin isoform (Makarenko et al. 2004; Schoenauer et al. 2011; Pluess et al. 2015). The major changes that are observed in the subcellular organisation of the cardiomyocytes in DCM happen at the intercalated disc. We consistently see an upregulation in the expression of actin-anchoring proteins such as adherens junction components (beta-catenin, cadherin) and also vinculin and N-RAP (Ehler et al. 2001; Pluess et al. 2015). This is potentially a response to an increased convolution of the plasma membrane and is also accompanied by a more intense signal for filamentous actin. Whether the elevated levels of an actin polymerising factor, the formin FHOD1, which are observed at the intercalated disc in DCM cause this increase in actin remains to be demonstrated (Dwyer et al. 2014). In mouse models for DCM this altered adherens junction composition is accompanied by a reduction in the numbers of gap junctions that are detected, suggesting impaired intercellular communication (Ehler et al. 2001). This observation can only be reproduced in a subset of the human patient material, indicating a difference between mouse and human phenotype and a potential compensatory effect by differential medication of human patients.
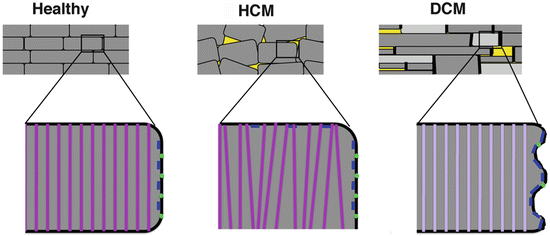
Fig. 1.4
Hypertrophic cardiomyopathy (HCM) is characterised mainly by myocyte disarray while dilated cardiomyopathy (DCM) is characterised by a general loss of size control, alterations in intercalated disc composition and by a switch to the expression of more elastic isoforms of the structural proteins titin and myomesin. Schematic representation of heart tissue from control, HCM or DCM patients. Apart from areas of cell loss and fibrosis (yellow), there is usually no dramatic myocyte disarray seen in DCM, which is in contrast to HCM. The cardiomyocytes display more variability in their sizes and can have more irregular shapes. At the cellular level, HCM is characterised by myofibril disarray and lateral cell–cell contacts (shown in blue), while DCM shows a relatively mild myofibrillar phenotype with just changes in isoform expression (light purple representing EH-myomesin containing M-bands), but has its major alterations in intercalated disc phenotype and composition. Gap junction expression (green dots) is reduced in DCM in the mouse
1.3 How to Study Cardiac Cytoarchitecture
A multitude of studies have been carried out that investigate and compare the subcellular localisation of a variety of cytoskeletal proteins either in cardiomyocytes in culture or on heart sections using immunofluorescence with antibodies that specifically recognise a particular protein. Especially when combined with confocal microscopy, these investigations have told us a lot about the subcellular distribution of proteins that make up the cardiac cytoarchitecture during development and in disease. However, the researcher wants to go beyond these descriptive studies and to explore the role of different components in a more functional way. There the problem starts, because the ideal model system to study cardiac cytoarchitecture does not exist and what is available to us comes all with advantages and disadvantages (for a list comparing different systems see Table 1.1). Over the years many laboratories have used cultured cardiomyocytes for their analyses, which have the advantage that they retain some of their differentiated characteristics, for example they usually continue to beat rhythmically, but the obvious disadvantage that these cells are taken out of their environment and placed into a plastic dish in culture medium. This makes them extremely amenable for interference by addition of drugs and for microscopy, but also means that it cannot be excluded that some of the observations that are made are a culture artefact. Especially in the response to growth factors and also in the way myofibrils are assembled there seem to be dramatic differences between two-dimensional as in the petri dish and three-dimensional arrangements (Armstrong et al. 2000; Ehler et al. 1999). Another potential pitfall is that the stalwart system of cardiomyocytes in vitro are neonatal rat cardiomyocytes (NRC), which means that they are taken from an animal at a time when full maturity is not reached, yet. It is therefore entirely possible that the responsiveness to stimuli will be different compared to adult cardiomyocytes and their cytoarchitecture is definitely distinct (Rothen-Rutishauser et al. 1998). A lot of isoforms and proteins that are specific for either the embryonic or the adult heart are in the process of being switched around birth; therefore embryonic markers such as ANF expression and EH-myomesin isoform expression are still maintained at least in a subset of the NRC (Fig. 1.5a), while markers of sarcomere maturity such as the expression of telethonin or M-protein are not yet present in all the cells. In addition, a beating frequency in the hundreds per minute makes them dramatically different from the beating rate that would be expected from a human cardiomyocyte. Cardiomyocytes that were differentiated from human inducible pluripotent stem cells may become the method of choice for in vitro studies in the future, but are at the moment characterised by their costliness and also immature features as far as their physiology and cytoarchitecture is concerned, which makes them similar to differentiated human embryonic stem cells (Földes et al. 2014). Another inherent problem with cultured cardiomyocytes whether rodent or human is that their cytoarchitecture is different compared to in situ, especially as far as the cell–cell contacts are concerned. While in the postnatal heart cell–cell contacts would only be found where myofibrils terminate, they can also be situated laterally to myofibrils in cultured cardiomyocytes (see arrow in Fig. 1.5b). This means that the terminology intercalated disc-like structure would be more appropriate in this situation since while they contain all the major components, their ultrastructural architecture will be different to an intercalated disc in situ. Freshly isolated cardiomyocytes from adult mouse or rat hearts can only be obtained by retrograde perfusion of the tissue with collagenase using a Langendorff apparatus and are characterised by a cytoarchitecture that is as close to the tissue as possible. However, they only have a limited life span to do experiments on them, which is even shorter for the ones from mouse than for the ones from the rat heart. The dedifferentiation process starts immediately and for example the specialised plasma membrane organisation is lost, with slightly different dynamics between rat and mouse (Pavlovic et al. 2010). A possibility to maintain the characteristic subcellular distribution of proteins such as connexin-43 a bit longer may be to assemble the adult cardiomyocytes to three-dimensional spheres using gravity (Kelm et al. 2006).




Table 1.1
Model systems to study cardiac cytoarchitecture
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
