Fig. 12.1
The ubiquitin-proteasome system and steps in the ubiquitylation and degradation of cellular substrate proteins. The ubiquitin and 26S proteasome structures were adopted from Vijay-Kumar et al. (1987) and Unverdorben et al. (2014), respectively. Specific proteasome subunits mentioned in the text are highlighted. The box contains, besides the structure of ubiquitin, a phylogenetic tree diagram of ubiquitin-like modifiers found in mammals
While substrate proteins may be released from the E3-ligase once they have undergone mono-ubiquitylation, marking them for degradation requires the formation of poly-ubiquitin chains. This poly-ubiquitylation is possible by the occurrence of seven lysine residues within the ubiquitin protein itself, such as lysine 48 (K48) or lysine 63 (K63), which can be utilized for attachment of another ubiquitin, and results in the formation of serially linked ubiquitins (also called poly-ubiquitin chains). Several models exist that explain how E3-enzymes promote ubiquitin chain formation (reviewed in Hochstrasser 2006). The most prominent of these models features the sequential addition of a single ubiquitin to a growing ubiquitin chain on the substrate protein. HECT-type E3-ligases, however, may first assemble the poly-ubiquitin chain onto a ubiquitin that is linked through a thioester bond to a cysteine within the enzyme and then transfer it onto the substrate protein (Verdecia et al. 2003). In addition, some E2-enzymes can assemble poly-ubiquitin chains without the aid of E3-enzymes (Chen et al. 1991), or poly-ubiquitin chain generation may be facilitated by formation of E2-enzyme homo- or heterodimers (Chen et al. 1993; Varelas et al. 2003).
Several factors have been reported that influence the ubiquitin chain type (e.g., K48-linked vs. K63-linked). These include pseudo E2-enzymes (also called ubiquitin E2-variant proteins) that resemble E2-enzymes but lack the catalytically active cysteine for ubiquitin thioester bond formation (Moraes et al. 2001; VanDemark et al. 2001). Other factors comprise conformational side-chain arrangements in E2-enzymes (Petroski and Deshaies 2005) or simply E3-ligase specificity that favors one type of poly-ubiquitin linkage over another (reviewed in Dye and Schulman 2007).
Mono-ubiquitylation of substrates, or modification of substrate proteins with poly-ubiquitin chains other than K48-linked, is typically associated with signaling events (e.g., DNA damage, transcriptional activation) or cellular trafficking (reviewed in Chen and Sun 2009; Shih et al. 2000). Typically, only substrates that have been modified by K48-linked poly-ubiquitin chains are routed for degradation through the 26S proteasome (Chau et al. 1989; van Nocker and Vierstra 1993). However, newer research indicates that some substrates modified by K63-linked ubiquitin chains may be also routed to the 26S proteasome (Saeki et al. 2009) or via p62/SQSTM1 to another cellular degradation system, the autophagy/lysosome system (Pankiv et al. 2007; Seibenhener et al. 2004). This specificity of the 26S proteasome for poly-ubiquitylated substrates modified by K48-linked (and K29-linked) ubiquitin chains is achieved through the essential Rpn10/S5a/PSMD4 and Rpn13/ADRM1 subunits, located in the 19S regulatory particle of the proteasome (Deveraux et al. 1994; Hamazaki et al. 2007; Husnjak et al. 2008; Peth et al. 2010; Zhang et al. 2009). Although biochemical studies indicated that Rpn10 is able to interact with K6-, K11-, K29-, and K48-linked poly-ubiquitin chains (Baboshina and Haas 1996; van Nocker et al. 1996), only poly-ubiquitin chains linked via K11, K29, K48, and K63 have been observed in vivo (Arnason and Ellison 1994; Chau et al. 1989; Spence et al. 1995; van Nocker and Vierstra 1993; Xu et al. 2009). After substrate recognition by the (poly-)ubiquitin receptors Rpn10 and Rpn13, the 19S particle undergoes extensive conformational changes that lead to substrate de-ubiquitylation and translocation of the substrate protein towards the 20S core particle. De-ubiquitylation (DUB) is achieved by one of the three proteasome subunits that possess DUB-function: Rpn11/PSMD14, Rpn13/Uch37/ADRM1, and Ubp6/Usp14 (reviewed in Finley 2009; Voges et al. 1999). While all these enzymes have de-ubiquitylation activity, their processivity differs: Rpn11 activity is ATP dependent, Rpn13 releases mono-ubiquitin, while Ubp6 was shown to release di- and tri-ubiquitin from poly-ubiquitin chains (Hanna et al. 2006; Lam et al. 1997). Following binding of poly-ubiquitylated substrates to ubiquitin receptors Rpn10 and Rpn13, and the processing of the poly-ubiquitin chain by proteasome subunits, the proteasome becomes committed to substrate degradation in an ATP-dependent step. This commitment step involves the recognition of unstructured regions in the substrate (Prakash et al. 2009) and activity of the six AAA-ATPase subunits (Rpt1–6/PSMC1–6) of the 19S proteasome base (Peth et al. 2010). The ATPases unfold globular substrate proteins and translocate the polypeptide chain towards the 20S core particle of the proteasome (Smith et al. 2007). The commitment of the proteasome towards a poly-ubiquitylated substrate was also shown to allosterically open a gate in the 20S core particle, which allows for entry of the unfolded polypeptide (Bech-Otschir et al. 2009; Li and Demartino 2009; Schreiner et al. 2008). The core particle consists of four ring-like multi-protein complexes (two heptameric alpha- and two heptameric beta-subunits) that form a central channel in which the unfolded polypeptide chain of the substrate translocates into (reviewed in Kunjappu and Hochstrasser 2013). Alpha subunits of the 20S core interact with proteins in the 19S regulatory particle to control gate opening, while the beta-subunits, specifically beta1, beta2, and beta5, harbor the caspase-like, trypsin-like, and chymotrypsin-like enzymatic activity to proteolytically cleave the substrate polypeptide into smaller fragments (reviewed in Tanaka 2013).
12.1.2 Autophagy/Lysosome
Eukaryotic cells developed another pathway for the removal of less degradable proteins, large molecular complexes, and even cellular organelles: the autophagy/lysosome system. Autophagy is commonly divided between directed or “selective” autophagy of specific substrates and so-called macro- or nonspecific autophagy of larger complexes, macromolecules, or cellular organelles, such as mitochondria (autophagy of mitochondria is termed “mitophagy”). The substrate is in either case encapsulated in a double-membraned vesicle called the autophagosome. Many of the studies that identified functions of autophagy-related genes/proteins (also abbreviated as ATG or APG) have been characterized in yeast (Klionsky et al. 2003); however, they are readily translatable to higher organisms, such as mammals, due to their high degree of evolutionary conservedness (Hale et al. 2013; Mizushima et al. 2011).
Degradation of substrates through the autophagy/lysosome system starts with the small protein LC3 (Map1LC3/ATG8) in an activation cascade that involves E1-like activating, E2-like conjugating, and E3-like lipidating enzymatic reactions. These reactions are similar to the canonical ubiquitylation cascade (Figs. 12.1 and 12.2). Moreover, a second enzymatic cascade involving the related ATG12 protein (Fig. 12.2 and box in Fig. 12.1) is required to assemble the ATG12-ATG5 E3-like lipidating enzyme for LC3/ATG8 (Ohsumi 2001).
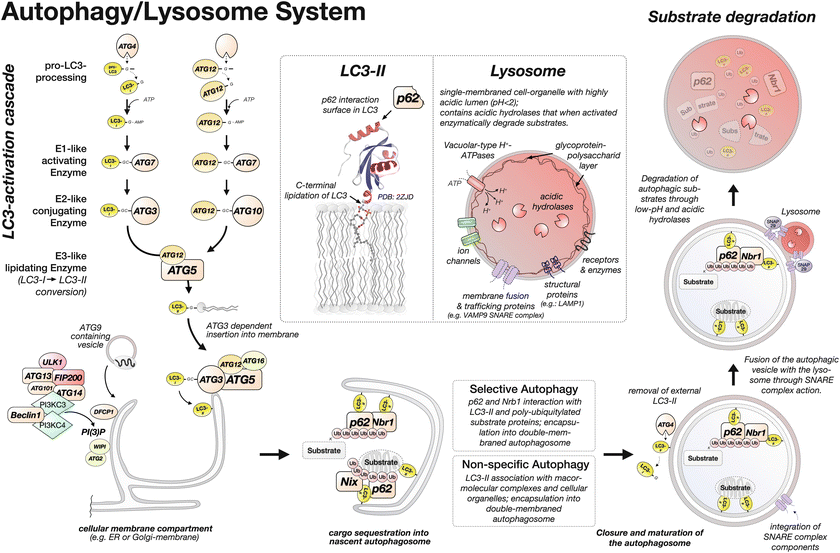
Fig. 12.2
The autophagy/lysosome system and steps in the degradation of cellular substrates. The structure of LC3/ATG8 was adopted from Ichimura et al. (2008)
Both LC3/ATG8 and ATG12 are proteolytically processed to enable the thioester bond formation between the C-terminal glycine residue in LC3/ATG8 or ATG12 and a cysteine residue on the E1-like activating enzyme ATG7. The proteolytic cleavage of the pro-LC3/ATG8 C-terminus is done by one of four ATG4 cysteine proteases (Kirisako et al. 2000), with ATG4B displaying the broadest processivity that extends to the closely related Gabarap and Gabarap-like family proteins (Gabarapl1, Gabarapl2/GATE16, and Gabarapl3; box in Fig. 12.1) (Kumanomidou et al. 2006; Li et al. 2010; Tanida et al. 2006). Once proteolytically processed, the LC3/ATG8 or ATG12 C-terminal glycine residue is adenylated in an ATP-dependent step and then coupled onto a cysteine residue in ATG7 through thioester bond formation. ATG7 works somewhat differently from the “canonical” ubiquitin E1-activating enzyme Uba1. LC3/ATG8- or ATG12-bound ATG7 forms homodimers (Komatsu et al. 2001). Once coupled to the ATG7 homodimer, LC3/ATG8 is transferred onto a cysteine residue in the E2-like conjugating enzyme ATG3, in a trans-thiolation reaction (Noda et al. 2011). A similar mode of action has been proposed for the thioester transfer reaction of ATG7-bound ATG12 onto ATG10 (Kaiser et al. 2013). ATG12 is finally irreversibly transferred onto ATG5 (Mizushima et al. 1998) through isopeptide bond formation and forms with the help of ATG16 a multimeric ~350 kDa ATG12-ATG5 complex (Kuma et al. 2002; Mizushima et al. 1999, 2003; Parkhouse et al. 2013). This multimeric ATG12-ATG5 complex now serves as E3-like enzyme in the transfer of LC3/ATG8 from ATG3 onto a phospholipid, phosphatidylethanolamine (PE) (Hanada et al. 2007; Ichimura et al. 2000). Newer research unveiled that the lipidation reaction proceeds in an ATG3-dependent fashion most efficiently at strongly deformed membrane structures present on the forming autophagosome rim (Nath et al. 2014) (Fig. 12.2). The un-lipidated and lipidated versions of LC3/ATG8 are sometimes referred to as LC3-I and LC3-II, respectively, and can be detected on immunoblot gels due to a slight shift in their molecular weights (Kabeya et al. 2004). Similarly, the closely related Gabarap and Gabarap-like proteins were also shown to exist in two versions: form I and form II, representing the un-lipidated and lipidated ubiquitin-like proteins, respectively.
Formation of the nascent autophagosome (sometimes also referred to as phagophore or omegasome) is thought to involve the endoplasmic reticulum membrane, although other cellular membranes, such as the plasma membrane, the membrane of the Golgi apparatus, or the outer mitochondrial membrane, have been suggested as autophagosome origin (Axe et al. 2008; Hailey et al. 2010; Moreau and Rubinsztein 2012; Yen et al. 2010). The autophagosome formation is thought to be initiated by a starvation signal originating from the mTORC1 pathway that acts on a protein complex consisting of the uncoordinated 51-like kinase 1 (ULK1/ATG1), ATG13, ATG14, ATG101, and FIP200/ATG17 (Hara et al. 2008; Itakura and Mizushima 2010; Karanasios et al. 2013; Kim et al. 2011; Mercer et al. 2009). The activated complex localizes to the nascent autophagosome (Hosokawa et al. 2009; Jung et al. 2009). Another signal for autophagosome initiation is connected to phosphatidylinositol 3-phosphate PI(3)P formation by class III phosphoinositide 3-kinase complexes, consisting of PIK3C3/Vps34, PIK3R4/p150/Vps15, Beclin1/ATG6, and ATG14 (Kihara et al. 2001; Suzuki et al. 2007; Tassa et al. 2003). Newly generated PI(3)P attracts another protein complex consisting of WIPI (WD40-repeat domain phosphoinositide-interacting protein; ATG18 analogs) family proteins and ATG2, as well as DFCP1 (Axe et al. 2008; Itakura and Mizushima 2010; Polson et al. 2010; Proikas-Cezanne et al. 2004) (Fig. 12.2). Finally ATG9, a transmembrane protein localized in endosomes, is thought to provide the nascent autophagosome with membrane vesicles (Mari et al. 2010; Orsi et al. 2012; Yamamoto et al. 2012).
Lipidated LC3-II/ATG8 in the nascent autophagosome membrane is then able to attract substrates for degradation. This feat is achieved by interacting with adaptor proteins that anchor the substrates to the lipidated LC3-II/ATG8. Examples of LC3-II/ATG8 interacting adaptor proteins are p62/SQSTM1, Nbr1, or NIX. p62/SQSTM1 and Nbr1 serve as adaptor proteins for poly-ubiquitylated substrates (Bjorkoy et al. 2005; Kirkin et al. 2009; Lamark et al. 2009; Pankiv et al. 2007). NIX resides in the mitochondrial membrane and plays an essential role for mitophagy (Ding et al. 2010; Novak et al. 2009). Encapsulation of substrates and closure of the autophagic vesicle may require actions of ATG4 proteases, presumably to remove LC3-II/ATG8 from the external membrane, and allow for autophagosome maturation (Betin et al. 2013; Yu et al. 2012). After encapsulation and closure, the mature autophagosome merges with the lysosome through SNARE complex-mediated membrane fusion (Itakura et al. 2012). Syntaxin-17, a SNARE complex protein, is specifically incorporated into membranes of mature autophagosomes and was recently shown to interact with SNAP29 and VAMP9, localized in lysosomal membranes. After lysosome fusion, the inner membrane will dissolve and the acidic environment as well as acidic hydrolases and proteases (e.g., cathepsins) from the lysosome will degrade macromolecular complexes, proteins, and membranes (Koike et al. 2005; Tatti et al. 2012).
12.1.3 Proteases
While protease functions are associated with the ubiquitin-proteasome system (e.g., de-ubiquitylating enzymes, beta-subunits of the proteasome) and the autophagy/lysosome system (ATG4 cysteine proteases, cathepsin proteases of the lysosome), “free” cellular proteases have also been shown to play important functions for signaling and clearance of substrates. Proteases are enzymes that hydrolyze peptide bonds within proteins. While comparative genomic and proteomic approaches identified more than 500 proteases in mammals, their actions, substrate specificity, and tissue-specific expression vary widely (Puente et al. 2003) (Table 12.1, Fig. 12.3). Proteases can be classified into five groups according to their mechanism of action: aspartic, metallo-, serine, threonine, and cysteine proteases. Aspartic and metalloproteinases require an activated water molecule in their active site for a nucleophile attack on the peptide bond in the substrate protein. In the other proteases, specific serine, threonine, or cysteine residues in the active site are utilized for the nucleophilic attack on the peptide bond, which gave rise to their respective names (serine, threonine, or cysteine proteases).
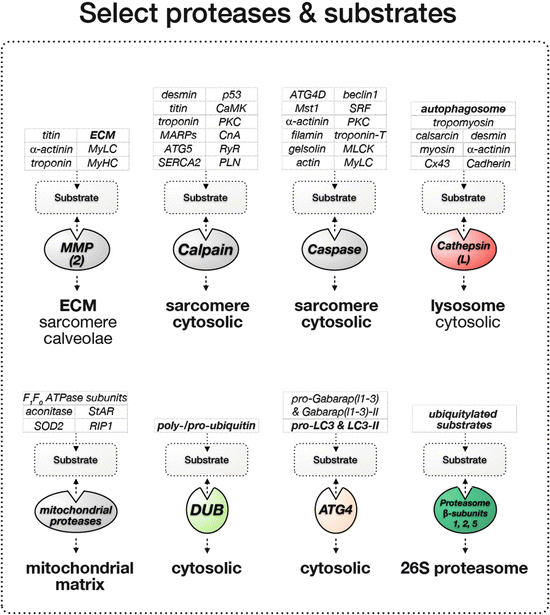
Table 12.1
Select proteases and known cardiac substrates
Protease | Select potential and confirmed substrate | Disease link |
|---|---|---|
Matrix metalloproteinases (MMP2) | ECM Alpha-actinin (Sung et al. 2007) Troponin-I (Wang et al. 2002) Titin (Ali et al. 2010) Myosin light chain (Sawicki et al. 2005) Myosin (Rouet-Benzineb et al. 1999) | Cardiac remodeling in MMP2 overexpressing mice (Bergman et al. 2007) Cardiac remodeling, fibrosis, inflammation (Passino et al. 2014) Angiogenesis, heart development, ischemia (Luttun et al. 2000) Heart failure (Muller and Dhalla 2011) |
Cathepsins (cathepsin-L) | Calsarcin (Petermann et al. 2006) Desmin (Petermann et al. 2006) Tropomyosin (Petermann et al. 2006) Alpha-actinin (Sun et al. 2013) Myosin (Sun et al. 2013) Connexin-43 (Sun et al. 2013) H-cadherin (Sun et al. 2013) | Cardiac hypertrophy, contractile dysfunction (Hua et al. 2012) Heart failure (Muller and Dhalla 2011) Cardiac inflammation, autophagosome accumulation (Pan et al. 2012) Cardiomyocyte apoptosis (Roberg and Ollinger 1998) |
Calpains | Troponin-T (Ho et al. 1994) Myosin (Pemrick and Grebenau 1984) Nebulin (Taylor et al. 1995) Dystrophin (Yoshida et al. 1992) Filamin (Davies et al. 1978) Ankyrin-B, -R (Harada et al. 1997) Beta-catenin (Li and Iyengar 2002) Talin (Muguruma et al. 1995) Vinculin (Taylor et al. 1995) Calcineurin (CnA) (Lakshmikuttyamma et al. 2004) SERCA2 (Singh et al. 2004) Phospholamban (PLN) (Singh et al. 2004) RyR (Singh et al. 2004) p53 (Kubbutat and Vousden 1997) PKCalpha (Kang et al. 2010) PKCdelta (Yamakawa et al. 2001) PKCepsilon (Saido et al. 1992) MyLCK (Ito et al. 1987) | Limb-girdle muscular dystrophy LGMD2A (Kramerova et al. 2008; Ojima et al. 2010; Richard et al. 1995) Dilated cardiomyopathy in calpastatin transgenic mice (Galvez et al. 2007) Increased fibrosis, sarcolemmal defects in calpain 4 knockout hearts (Taneike et al. 2011) Heart failure (Muller and Dhalla 2011) Cardiovascular disease (Sorimachi and Ono 2012) |
Caspases | Myosin light chain (MyLC) (Moretti et al. 2002) Myosin light-chain kinase (Petrache et al. 2003) Gelsolin (Martin et al. 2010) Alpha-actinin (Communal et al. 2002) PKC-δ (Narula et al. 1999) mammalian sterile 20-like kinase 1 (Mst1) (Yamamoto et al. 2003) Serum response factor (SRF) (Chang et al. 2003) ATG4D (Betin and Lane 2009) Beclin1 (Wirawan et al. 2010) | Roles of caspases in: Heart failure (Muller and Dhalla 2011) |
ATG4 | Pro-LC3 and delipidation of LC3-II Pro-Gabarap(l1-3) and delipidation of Gabarap(l1-3)-II | |
DUBs (de-ubiquitinating cysteine proteases) | Poly-ubiquitin chains Pro-ubiquitin | |
Mitochondrial proteases (Lon protease) | SOD2 (Bayot et al. 2010) RIP1 (Bayot et al. 2010) mtHSP60 (Bayot et al. 2010) F1F0-ATPAse subunit 1 and 2 (Bayot et al. 2010) StAR (Bayot et al. 2010) Aconitase (Bota and Davies 2002) | Pressure overload-induced cardiomyopathy (Hoshino et al. 2014) Friedreich’s ataxia (Guillon et al. 2009) |
Fig. 12.3
Select proteases and cardiac substrates
12.1.3.1 Matrix Metalloproteinases
The matrix metalloproteinase (MMP) system plays important functions for degradation and remodeling of the extracellular matrix in hearts (Liu et al. 2006). These calcium- or zinc-dependent enzymes are able to degrade most extracellular proteins, like collagen (MMP1, 8, 13, and 18), elastin, laminin, or fibronectin (e.g., MMP3, 7, 10), and have been shown to play critical roles for cardiac remodeling after myocardial infarction or in fibrosis (for a review see Hua and Nair 2015; Luttun et al. 2000; Passino et al. 2014). MMPs are expressed as inactive proenzymes (zymogens), which become active after proteolytic cleavage (Massova et al. 1998). While most substrates for MMPs are located in the extracellular space, intracellular functions for MMP2 have been reported on sarcomeric proteins, such as alpha-actinin, troponin-I, titin, or myosin light and heavy chains (Ali et al. 2010; Rouet-Benzineb et al. 1999; Sawicki et al. 2005; Sung et al. 2007; Wang et al. 2002) (Table 12.1). Indeed, transgenic overexpression of MMP2 leads to cardiac remodeling, diastolic dysfunction, and development of dilated cardiomyopathy (Bergman et al. 2007). The action of MMPs can be regulated by the four members of the tissue inhibitor family of matrix metalloproteinases (TIMPs) (Woessner 1991). While all cardiac cell types, including cardiomyocytes, have been demonstrated to express MMPs, cardiac fibroblasts and endothelial cells constitute the main sources for MMPs and TIMPs in the heart under basal conditions (Coker et al. 1999; Tyagi et al. 1995). It was also demonstrated that during cardiac remodeling infiltrating inflammatory cells (neutrophiles, macrophages) may contribute additional MMPs (Romanic et al. 2002). Newer research indicated that besides TIMPs, MMP2 action in cardiomyocytes, cardiac fibroblasts, and endothelial cells can be inhibited by the scaffolding domain of caveolins 1 and 3 (Chow et al. 2007; Kandasamy et al. 2009) as well as by inhibitors of other proteases, such as calpastatin (Kandasamy et al. 2009).
12.1.3.2 Cathepsins
Cathepsins are proteases that can be found in the lysosome and contribute to autophagic degradation of cellular substrates. Cathepsins can be classified either as serine, aspartic, or cysteine proteases (Hua and Nair 2015). Recent studies also described cathepsin action in neutral conditions, such as the cytosol of cells (Sever et al. 2007). Specifically, cathepsin-L has been shown to play important roles for the heart, as mice lacking this protease develop dilated cardiomyopathy with accumulation of lysosomes, mitochondrial degeneration, and aggregation of sarcomeric proteins alpha-actinin and myosin, as well as higher levels of connexin-43 and H-cadherin (Petermann et al. 2006; Spira et al. 2007; Stypmann et al. 2002; Sun et al. 2013) (Table 12.1). Other cathepsins, such as cathepsin-K, -S, or -D, have also been demonstrated to play roles during hypertrophy, contractile dysfunction, cardiac inflammation, or oxidative stress (Hua et al. 2012; Pan et al. 2012; Roberg and Ollinger 1998).
12.1.3.3 Calpains
Calpains are a large family of calcium-dependent papain-like cysteine proteases. Calpain proteases can be classified according to their calcium requirements into m-calpains, which require millimolar amounts of calcium for their activity, and μ-calpains that work in the presence of micromolar amounts of calcium (Cong et al. 1989). Further classification of the 15 mammalian calpains can be done as specified by their domain layout into typical-calpains, which possess a C-terminal calmodulin-like domain and atypical-calpains that lack this domain (Sorimachi and Ono 2012; Suzuki et al. 1995). Lately, calpain nomenclature has been unified according to the gene products defined by the Human Genome Organization Gene Nomenclature Committee (Sorimachi et al. 2011).
Despite the ubiquitous expression of many calpains, some of them show distinctive tissue-specific expression patterns, like calpain-3, which is prominently expressed in skeletal muscle cells and, when mutated, can be the molecular cause for a form of muscular dystrophy (Kramerova et al. 2008; Ojima et al. 2010, 2011; Richard et al. 1995). Although mainly expressed in skeletal muscles, calpain-3 may also play a role in the heart (Fougerousse et al. 2000; Sarparanta et al. 2010; Taveau et al. 2002). Calpains 1 and 3 have been shown to localize to the myofibrils at the M-band and I-band of the sarcomere, indicating specific roles for these proteases at these sites (Hayashi et al. 2008; Raynaud et al. 2005). Targets for calpains in muscle include titin and titin-binding proteins of the muscle ankyrin repeat domain-containing protein (MARP) family (Hayashi et al. 2008; Laure et al. 2010; Suzuki et al. 1996), troponin-I (Maekawa et al. 2003; van der Laarse 2002), desmin (Galvez et al. 2007), calcineurin (CnA) (Lakshmikuttyamma et al. 2004), SERCA2-ATPase, phospholamban (PLN), ryanodine receptor (RyR) (Singh et al. 2004), calmodulin-dependent protein kinase (CaMK) (McGinnis et al. 1998), p53 (Kubbutat and Vousden 1997), or PKCalpha (Kang et al. 2010) (Table 12.1). Although the variety of substrates for calpains render it difficult to find conserved proteolytic motifs, targeted sequences are usually intrinsically unstructured and show increased abundance of proline, glutamic acid, serine, and threonine residues, similar to PEST sequences that were found to be susceptible for degradation by the ubiquitin-proteasome machinery (Shumway et al. 1999).
Several factors have been shown to regulate calpain activity. These are in addition to calcium, calpastatin (Murachi 1989), and phosphorylation by protein kinases, such as the extracellular regulated kinase (ERK) (Inserte et al. 2009).
Calpain function is required for cardiac development and cardiac function. Mice that overexpress calpain inhibitor calpastatin, causing a 50 % reduction of calpain activity, develop dilated cardiomyopathy (Galvez et al. 2007). Moreover, conditional knockout mice for calpain 4, a regulatory subunit for calpains 1 and 2, displayed increased fibrosis and sarcolemmal defects under hemodynamic stress, indicating important functions for these proteases in cardiac adaptation and remodeling (Taneike et al. 2011).
12.1.3.4 Caspases
Caspases are cysteine proteases that are best known for their role during signaling and cell apoptosis (Stephanou et al. 2001; Yuan et al. 1993, reviewed in Chowdhury et al. 2008). Similar to matrix metalloproteinases, caspases exist first as inactive form (precursor zymogen), before becoming active through a proteolytic cleavage event that separates the tripartite proenzyme. Further classification of caspases can be done depending on their function into group I caspases that play a role for inflammatory responses (caspases 1, 4, 5, 12–14), group II caspases that initiate apoptosis (caspases 2, 8–11), and group III caspases that are also called effector or executioner caspases (Caspases 3, 6, 7) (Deveraux et al. 1998). Activation of caspases is achieved through a proteolytic cleavage event after conserved aspartic acid residues either by autoactivation or through the action of other caspases (Yang et al. 1998, reviewed in Chowdhury et al. 2008). After processing, caspases assemble into a hetero-tetramer consisting of two large subunits that each contains a catalytic protease site and two smaller subunits that structurally link the complex together (Yang et al. 1998).
As caspase activity is linked to apoptosis and cell death, most investigations into the role of caspases for the heart have been done in various forms of cardiomyopathy (Birks et al. 2008; Ghosh et al. 2005; Sharma et al. 2007), myocardial infarction (Olivetti et al. 1996; Piro et al. 2000; Zidar et al. 2006), and ischemia-reperfusion (Fliss and Gattinger 1996; Gottlieb et al. 1994; Kang et al. 2000). Besides activating the cellular apoptotic signaling cascade, caspases may also directly cleave myofibrillar substrates, as injection of active caspase 3 into healthy cardiomyocytes rapidly degraded sarcomeric structures (Laugwitz et al. 2001). Indeed, potential substrates include actin (Maravei et al. 1997; Mashima et al. 1999), myosin light chain (MyLC) (Moretti et al. 2002), myosin light-chain kinase (Petrache et al. 2003), gelsolin (Martin et al. 2010), troponin-T (Communal et al. 2002; Lancel et al. 2005), filamin (Browne et al. 2000; Umeda et al. 2001), or alpha-actinin (Communal et al. 2002) (Table 12.1).
12.1.3.5 Mitochondrial Proteases
While degradation of whole mitochondria within cells occurs preferentially through the autophagy/lysosome system (mitophagy), mitochondria developed a protein quality control and degradation system to manage misfolded or oxidatively damaged and denatured proteins (Wang et al. 1993). The best characterized system involves actions of the Pim1/Lon and Clp proteins, mitochondrial proteases that localize to the mitochondrial matrix (Kang et al. 2002; Suzuki et al. 1994). Both proteases have been identified in most organisms and show a remarkable degree of evolutionary conservedness. Lon protease contains in addition to its protease domain an AAA-ATPase domain, which is important for target selection, unfolding, and translocation of the substrate protein towards the protease domain (similar to AAA-ATPase subunits of the 26S proteasome) (Ondrovicova et al. 2005). The structure of ClpP proteases was found to be remarkably similar to the beta-subunits of the 26S proteasome, forming a doughnut-shaped heptameric ring structure (de Sagarra et al. 1999; Kessel et al. 1995).
While degradation signals, such as poly-ubiquitylation, have not been identified for substrate proteins of mitochondrial proteases, Lon protease was shown to specifically degrade the oxidized (damaged) version of aconitase over the functionally active native version of the enzyme (Bota and Davies 2002). Aconitase is an essential mitochondrial enzyme that is particularly susceptible to oxidative damage and has been demonstrated to be of importance for cardiac function and in disease (Bota and Davies 2002; Huang et al. 2001; Lin et al. 2009; Rotig et al. 1997; Yan et al. 1997). It has been shown for yeast that the activities of mitochondrial proteases, such as Lon, in combination with chaperones, protect this cell organelle from protein aggregation (Bender et al. 2011). Other substrates for mitochondrial proteases, such as Lon protease, may include SOD2, RIP1, mtHSP60, F1F0-ATP synthase subunits 1 and 2 (Bayot et al. 2010), or the steroidogenic acute regulatory protein (StAR), which is expressed in cardiac fibroblasts and may play a role for their survival (Anuka et al. 2013; Ondrovicova et al. 2005) (Table 12.1). The decrease in activity of this system during aging and in degenerative disease could be particularly harmful, as mitochondrial dysfunction may lead to an increase in cellular reactive oxygen species, triggering caspase activation, and result in cellular apoptosis (Bota et al. 2005; Delaval et al. 2004; Ngo and Davies 2007). Results from mice undergoing pressure overload-induced hypertrophy indicated inactivation of Lon-protease activity through its oxidation (Hoshino et al. 2014), further hinting at important roles of this class of proteases for cardiac maintenance. On the other hand, a mouse model for Friedreich’s ataxia shows upregulation of mitochondrial proteases Lon and ClpP that correlate with a loss of mitochondrial Fe-S proteins and progression of the disease (Guillon et al. 2009). Taken together, the data indicate that mitochondrial proteases are important regulators of mitochondrial homeostasis. More recent data suggest further that their function may extend beyond degradation of damaged proteins, as they bind to mitochondrial DNA and RNA (Liu et al. 2004), and may be involved in the replication of the mitochondrial genome.
12.1.4 Chaperones and Heat-Shock Proteins
Chaperones and heat-shock proteins serve multiple functions within cardiac cells. They form an important part of the ERAD system by detecting misfolded proteins, assist in the folding of newly generated polypeptides, aid in the integration and removal of proteins into larger macromolecular complexes (e.g., integration of myosin into the sarcomere), and protect proteins from aggregating. Each of these diverse functions requires a specialized subset of chaperones.
Well-characterized chaperones in cardiac muscles and fibroblasts are Hsp90, Hsp70, Unc45B, the family of HspB proteins, or Hsp47 (for a review see Nagata 1998; Willis et al. 2009b). Proteins of the HspB family (small heat-shock protein family, also called sHSP) with important cardiovascular functions are the muscle-specific HspB7/cvHSP (cardiovascular heat-shock protein), alphaB-crystallin/HspB5, and Hsp27/HspB2 or Hsp20/HspB6. Among these, alphaB-crystallin/HspB5 and Hsp27/HspB2 were shown to localize to the sarcomere (Lutsch et al. 1997; Yoshida et al. 1999). The heat-shock protein alphaB-crystallin/HspB5 reportedly binds to the titin N2B-region and protects this spring-like element from unfolding (Bullard et al. 2004). Knockouts for alphaB-crystallin/HspB5 and/or HspB2 demonstrated separate functions for the two chaperones, indicating that each of the HspB family members may exert special roles for muscles (Brady et al. 2001; Morrison et al. 2004; Pinz et al. 2008). Mutations in alphaB-crystallin/HspB5 have been shown to cause desmin-related myopathies and cardiomyopathies, such as dilated cardiomyopathy (Dalakas et al. 2000; Wang et al. 2001). Assembly of myosin into sarcomeres is aided by the functions of the striated muscle Unc45B, Hsp70, and Hsp90 (Barral et al. 2002; Srikakulam and Winkelmann 2004), which also display a sarcomeric localization pattern (Etard et al. 2008). While these chaperones were shown to have important functions for specific cardiac proteins (e.g., myosin or desmin), another heat-shock protein, Hsp27, was identified to have a more developmental role, suggested by its importance for heart tube formation (Brown et al. 2007). In addition, many cardiac chaperones and heat-shock proteins have been shown to be differentially modified and regulated in a variety of cardiomyopathies and during heart failure (Fan and Kranias 2010; Scheler et al. 1999; Stark et al. 2010).
Besides cardiomyocytes, heat-shock proteins play also important roles in cardiac fibroblasts and endothelial cells. The heat-shock protein Hsp47 forms part of the endoplasmic reticulum-associated degradation (ERAD) system in cardiac fibroblasts, where it transiently associates with procollagen to support the protein quality control. Therefore, Hsp47 functions play a crucial role during cardiac remodeling and fibrosis (Hagiwara et al. 2011; Nagata 1996, 1998).
12.1.5 Cooperativity Between Degradation Systems
It is important to point out that none of the degradation systems work in isolation but that there is in fact a high degree of cooperativity and cross talk between the UPS, autophagosome/lysosome system, cellular proteases, as well as chaperones. Examples for the regulation of cellular autophagy include the degradation of autophagy-related proteins ATG13 and ATG5 by the UPS (Hammerling and Gustafsson 2014; Mercer et al. 2009), the clearance of poly-ubiquitylated proteins via autophagy (Fan et al. 2010; Lamark et al. 2009; Novak et al. 2009; Seibenhener et al. 2004), or the regulation of autophagy by calpain1- and caspase-dependent cleavage of ATG5 (Xia et al. 2009; Yousefi et al. 2006) or beclin1 (Wirawan et al. 2010).
The activity of cellular proteases was shown to be dependent on the action of E3-ubiquitin ligases, as mono-ubiquitylation of caspases 3 and 7 has been demonstrated to regulate their activity (Huang et al. 2000). Cytoplasmic proteases and their inhibitors have also been shown to regulate matrix metalloproteinases (Kandasamy et al. 2009) and are important for the proteasomal degradation of a subset of myofibrillar proteins (Galvez et al. 2007).
12.2 Roles of Degradation Systems in the Heart
While the functions and mechanisms of the various cellular degradation systems are similar in all cell types, cardiomyocytes and cardiac fibroblasts show cell-type-specific adaptations to suit their specific needs and tissue-specific requirements.
12.2.1 Balance Synthesis/Degradation
Under physiological conditions, the heart mass is determined by the balance between muscle hypertrophy and muscle atrophy, which is directly linked to the regulation of protein synthesis and degradation. Deleterious diseases such as cardiomyopathies can result from defects in this balance, and comprehension of the underlying signaling pathways that influence protein synthesis and degradation is a field of intensive research (Heineke and Molkentin 2006; Razeghi and Taegtmeyer 2006). The increase in protein synthesis during hypertrophy largely contributes to the increase in cardiomyocyte size and the cardiac mass. The main signaling pathways underlying cardiac hypertrophy comprise, but are not restricted to, the activation of the calcineurin-NFAT pathway (Wilkins and Molkentin 2004), the calcium and calmodulin-dependent kinase II (CaMKII)-HDAC pathway (Swaminathan et al. 2012; Zhang et al. 2003; Zhu et al. 2000) that leads to activation of MEF2 (Wu et al. 2006), or the pathway encompassing insulin and insulin growth factor 1 (IGF1), PI3K, AKT, and GSK3-beta (Heineke and Molkentin 2006; Yu et al. 2010). Another way of promoting hypertrophy through the PI3K-AKT pathway includes activation of the mTORC1 protein complex. This complex is then able to promote protein synthesis through the activation of the p70/85 S6 kinase-1 (S6K1) and p54/56 S6K2, resulting in an increase of ribosomal biosynthesis (Heineke and Molkentin 2006).
During cardiac atrophy, the rate of protein degradation is predominant over the rate of protein synthesis. As a consequence, cell size and cardiac mass are decreased. The ubiquitin-proteasome system (UPS) plays a prominent role in atrophy, as UPS components like ubiquitin-B and UbcH2, as well as poly-ubiquitylated proteins, are generally found to be increased (Razeghi et al. 2006; Razeghi and Taegtmeyer 2006). The discovery of muscle-specific UPS components, such as E3-ligases of the MuRF family, or substrate adaptors for cullin-based E3-ligases, like atrogin1/MAFbx, triggered a multitude of investigations into molecular mechanisms, substrates, and roles during heart development and in disease.
12.2.2 Protein Quality Control and the ERAD System
Protein degradation is an important mechanism to maintain cardiac cell function. Indeed, misfolded proteins have been identified as molecular causes underlying the development of cardiac diseases. For example, production of misfolded alphaB-crystallin/HspB5 leads to crystallinopathies which include restrictive, hypertrophic, and dilated cardiomyopathies, as well as heart failure (Sanbe 2011). Another example are desminopathies (McLendon and Robbins 2011), where the accumulation of misfolded desmin is causative of the cardiomyopathy. Indeed, aberrant proteins need to be identified early during their synthesis or maturation and either refolded through the actions of chaperones or degraded.
The best characterized protein quality control mechanism is the endoplasmic reticulum-associated degradation (ERAD) system. Detection of abnormal proteins in the endoplasmic reticulum (ER) triggers the unfolded protein response (UPR). Misfolded proteins that are recognized by the ER trigger activation of chaperones, including the immunoglobulin heavy chain-binding protein (BiP), a lumenal Hsp70 protein (Brodsky 2007; Haas 1994), and the protein disulfide isomerase (PDI) (Molinari et al. 2002). Once identified, aberrant peptides are extracted from the ER through the retranslocon complex. This complex is constituted of membrane proteins assembled around E3-ubiquitin ligases, forming a pore in the membrane through which targeted proteins exit the ER (Carvalho et al. 2010; Zhang and Ye 2014). The translocation of proteins from the ER is believed to be highly energy dependent. While crossing the membrane, abnormal proteins undergo poly-ubiquitylation. A protein complex formed by p97/VCP [an AAA-ATPase protein (ATPases Associated with diverse cellular Activities)], Npl4, and Ufd1 was found to be required for the extraction of aberrant peptides from the ER (Zhang and Ye 2014). Once in the cytosol, the poly-ubiquitylated substrates are degraded by the 26S proteasome.
12.2.3 The Role of Substrate-Ubiquitylation Beyond Degradation
The ubiquitylation of proteins is a highly conserved posttranslational modification that serves versatile functions within cells. While the role for ubiquitin in protein degradation is now well characterized, its involvement in other biological pathways is emerging. There are numerous examples, where ubiquitylation of substrate proteins in cardiac cells modulates their functionality significantly.
Gap junctions are specialized plasma membrane domains enriched in connexin proteins that form channels (connexons) between adjacent cells. These channels are located at the intercalated discs (ID) and were found to be essential for cardiac action potential propagation, thus playing an important role in the intercellular communication (Kurtenbach and Zoidl 2014). As observed during injury, gap junctions are often and rapidly remodeled or disassembled. Indeed, the protein half-life of connexin-43 (Cx43), a major constituent of cardiac gap junctions, is thought to be between 1 and 3 h (Beardslee et al. 1998). While Cx43 has been shown to be degraded by the UPS and the lysosome, its mono-ubiquitylation may serve as an initial trigger that leads to clathrin-dependent internalization of the connexon into the cytoplasm, its disassembly, and ultimately poly-ubiquitylation (Leithe and Rivedal 2004a) (Fig. 12.4). This tightly regulated process is thought to be under control of several cellular signaling pathways and protein kinases (Lampe and Lau 2000; Leithe and Rivedal 2004a, b) and allows gap junctions to be quickly remodeled in response to cellular stress.
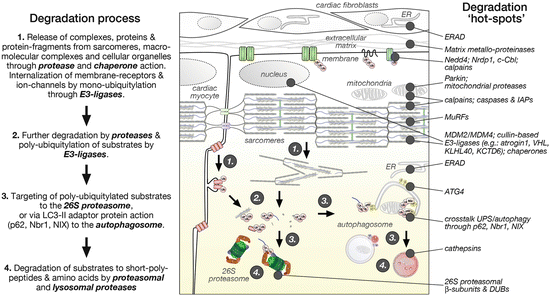
Fig. 12.4
The degradation process of cardiac proteins (adopted from Goll et al. 2003) and cellular “hot spots” for degradation pathway components
In a similar fashion to clathrin-mediated endocytosis of Cx43, caveolae are sites where cells internalize membranes, molecules, transmembrane, and membrane-bound proteins. Caveolin-1 (Cav1), -2, and -3 proteins are structural constituents of caveolae expressed in cardiomyocytes and were shown to play important roles in cardiac signaling and physiology (Panneerselvam et al. 2012; Yang et al. 2014). Interestingly, it was shown that ubiquitylation of the Cav1 N-terminus constitutes a signal for its internalization and sorting into lysosomes (Kirchner et al. 2013). Endo-lysosomal sorting of ubiquitylated Cav1 proteins was shown to be p97/VCP dependent, pointing out a new role for p97/VCP unrelated to ERAD (Ritz et al. 2011).
Endocytotic mechanisms for substrate internalization play also a role for cardiac ion channels. Indeed, major cardiac voltage-dependent channels are tightly regulated in a quantitative manner via endocytosis (Ishii et al. 2012). Ubiquitylation is a very common posttranslational modification observed on cardiac ion channels and is thought to control their removal from the sarcolemma (Rougier et al. 2010). Interestingly, members of NEDD4 E3-ligases that are enriched in cardiac tissue were shown to ubiquitylate various ion channels, such as the voltage-gated cardiac potassium channel KCNQ1 or hERG1 (Cui and Zhang 2013; Rougier et al. 2010).
Ubiquitylation of sarcolemmal ion channels constitutes therefore a rapid way to modulate the density of ion channels at the surface of the membrane of cardiomyocytes and may represent an important mechanism for controlling the duration of the cardiac action potential.
Another example for the role of protein-ubiquitylation beyond its function as a degradation tag are caspases, where mono-ubiquitylation of caspase 3 and 7 by E3-ligases of the IAP family has been shown to act as an intracellular targeting signal, which in addition modulates caspase activity (Huang et al. 2000).
Similarly, posttranslational modification of calmodulin (CaM) by ubiquitin is thought to modulate the functions of this intracellular calcium receptor and signaling protein (Berchtold and Villalobo 2013; Swulius and Waxham 2008). It was shown that CaM can be reversibly ubiquitylated by ubiquitin-calmodulin ligase in the presence of calcium in cardiac cells (Jennissen and Laub 1988; Ziegenhagen and Jennissen 1988). Interestingly, mono-ubiquitylated CaM was able to strongly decrease its signaling function and the activity of binding partners such as phosphorylase kinase (Laub et al. 1998). These studies illustrate that (mono-)ubiquitylation of CaM is not priming the protein for degradation by the UPS but is important for the modulation of its activity.
Mono-ubiquitylation of substrate proteins was also shown to be important for the regulation of transcription factors. Transcription factors of the FoxO-family (Forkhead box O) were demonstrated to be posttranslationally modified by the cullin1-atrogin1 E3-ligase via K63-linked poly-ubiquitin chains that led to their activation (Li et al. 2007a). Intriguingly, activation of FoxO transcriptional activity through this mechanism resulted in the expression of atrogin1, suggesting a feed-forward loop that may counteract the induction of cardiac hypertrophy.
12.2.4 Cardiac-Specific Adaptations of the Degradation Machineries
Sarcomeres are the basic contractile units of striated muscles. Therefore, their maintenance is crucial for the correct function of the muscles. Indeed, the discovery of UPS-related proteins localizing at the sarcomere is not surprising. These include de-ubiquitylating (DUB) enzyme complexes such as Abro1 (Cilenti et al. 2011), members of the ubiquitin-specific protease family USP17 (Shin et al. 2006), USP22 (Lee et al. 2006), USP2 (Gousseva and Baker 2003), or USP28 (Valero et al. 2001), as well as E3-ligases and their substrate adaptor proteins, like the muscle RING-finger proteins (MuRFs) or atrogin1.
Intriguingly, many of the muscle-specific, but also some of the more ubiquitously expressed degradation system components were found to be associated with the sarcomeres. Their subcellular localization may result from interaction partners and/or ubiquitylation substrates like titin, troponin, or myosin, which are integral parts of the sarcomeric apparatus. In addition, interaction of degradation machinery components with the sarcomere may regulate their function. Examples may include MuRF1 and other UPS components that interact with the titin M-band region. Degradation system components may also be sequestered to the sarcomere in order to “inhibit” their action under baseline conditions (e.g., calpain 3). Once activated, such as during cellular stress, their sarcomeric localization is lost.
This principle may also work in reverse, where cellular stress results in the sarcomeric association of proteins that are normally found in the cytoplasm (e.g., chaperones). Here stress on the myofilaments may result in the unfolding of sarcomeric components and trigger association of chaperones and other degradation pathway components.
12.2.5 The Cardiac Degradation Process
It is generally thought that chaperones and proteases release proteins and protein complexes from the myofilaments, other macromolecular complexes, or cellular organelles (Goll et al. 2003). These actions may be supported by E3-ligases, which were shown to regulate membrane-receptor and ion-channel internalization through mono-ubiquitylation. Protein substrates are then poly-ubiquitylated by substrate-specific E3-ligases for subsequent degradation by the UPS and/or sequestered into nascent autophagosomes for lysosomal degradation (Fig. 12.4).
The following paragraph highlights specific E3-ligases with known cardiac roles and discusses their involvement in cardiac development and for cardiomyopathies.
12.3 E3-Ligases with Cardiac Roles
12.3.1 MuRF Protein Family
The E3-ubiquitin ligase family of muscle RING-finger (MuRF) proteins consists of three highly related members: MuRF1/TRIM63/RNF28, MuRF2/TRIM55/RNF29, and MuRF3/TRIM54/RNF30. These muscle-specific E3-ligases are among the first ubiquitin-proteasome components that were shown to localize to the sarcomere in cardiac and skeletal muscle cells (Centner et al. 2001; McElhinny et al. 2004; Spencer et al. 2000). All members of the MuRF family display a similar domain layout, with an N-terminal RING domain, a centrally located BBox domain followed by coiled-coil domains, and a MuRF-specific unstructured region towards the C-terminus that may contain another motif, the COS-box (Short and Cox 2006). While MuRF2 was shown to express four different splice isoforms that are developmentally regulated in the heart (Perera et al. 2010), MuRF1 and MuRF3 are both expressed in adult striated muscles (including the heart) (Centner et al. 2001).
The N-terminally located RING domain in MuRF proteins, in combination with E2-conjugating enzymes of the UBCH5 family (Fielitz et al. 2007a; Kedar et al. 2004), is required for the ubiquitylation of substrates (Fig. 12.5). In addition it was shown that the RING domain may also interact with Sumo3, a small ubiquitin-like modifier (Dai and Liew 2001) (box in Fig. 12.1). This association of MuRF with the sumoylation machinery is further validated by interactions with the ubiquitin-conjugating enzyme 9 (Ubc9) as well as isopeptidase T-3 (ISOT-3); both enzymes are involved in the sumoylation cascade (McElhinny et al. 2002).
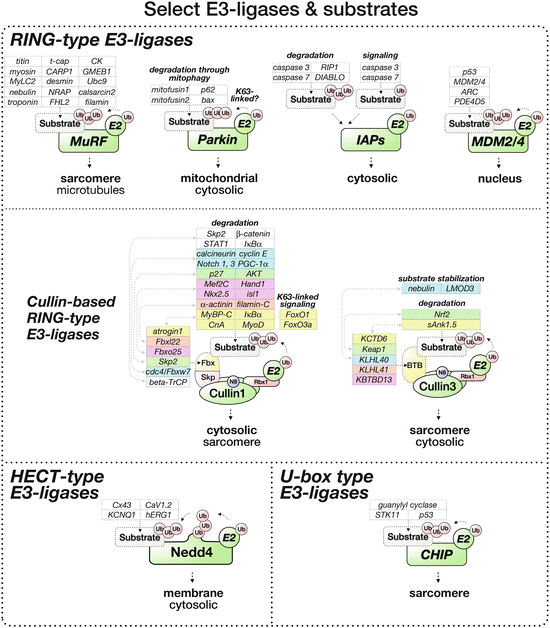
Fig. 12.5
Select E3-ligases and their cardiac substrates for ubiquitylation
The centrally located Bbox domain was shown to attract various interaction partners and potential substrates, including FHL2/DRAL, calsarcin-2, or CARP1/Ankrd1, a member of the muscle ankyrin repeat domain-containing protein (MARP) family (Witt et al. 2008) (Table 12.2 and Fig. 12.5 for more potential and verified MuRF substrates). The coiled-coil domains in MuRF proteins indicate the potential to form dimers. Indeed, the formation of homo- and heterodimers between all members of this family has been shown experimentally (Centner et al. 2001; Witt et al. 2005). While MuRF proteins display sequence variability in their C-terminal regions, this part of the protein contains a sequence motif called COS-box (also named acid-rich/AR region). This region was demonstrated to be responsible for the reported interaction and regulation of microtubules (Koyama et al. 2008; Short and Cox 2006), alongside the coiled-coil domains (Spencer et al. 2000).
Table 12.2
Potential and confirmed cardiac substrates for muscle RING finger (MuRF) E3-ligases
E3-ligase | Select potential and confirmed substrates | Disease link |
|---|---|---|
MuRF | Calsarcin-2 (Witt et al. 2008) Calcineurin-A (CnA) (Maejima et al. 2014) CARP1/Ankrd1 (Witt et al. 2008) Desmin (Witt et al. 2008) Troponin-T (Witt et al. 2005) Troponin-C (Kedar et al. 2004) Nebulette (Witt et al. 2008) NRAP (Witt et al. 2005) Vimentin (Rubel et al. 2013) p62 (Witt et al. 2008) mtHSP60/HSPD1 (Rubel et al. 2013) Adenylate kinase (Witt et al. 2005) Aldolase A (Witt et al. 2005) PIAS (Witt et al. 2008) Sumo-3 (Dai and Liew 2001) Ubc9 (McElhinny et al. 2002) ISOT-3 (McElhinny et al. 2002) MuRF1 (Rubel et al. 2013) | Muscle wasting/atrophy (Bodine et al. 2001; Kamalov et al. 2013; Moresi et al. 2010; Polge et al. 2012) Heart failure (Willis et al. 2009a) Aging (Witt et al. 2008) Myosin storage myopathy (Fielitz et al. 2007a) |
The best characterized sarcomeric interaction partners for MuRF1 are titin domains A168–A170, which are located adjacent to the stretch-sensitive protein kinase domain in titin (TK) (Lange et al. 2005; Mrosek et al. 2007; Muller et al. 2007). Indeed, transgenic mice for MuRF1 were found to display changes to the sarcomeric M-band, where this region of titin is located (Willis et al. 2009a). Moreover, the M-band region of titin may serve as a hot spot for the regulation of muscle gene expression and turnover, as MuRF2, p62/SQSTM1, and Nbr1 were also found to associate with titin-kinase in a stretch-dependent manner (Lange et al. 2005), and titin M-band knockout mice develop a severe form of atrophy (Peng et al. 2005). Both p62/SQSTM1 and Nbr1 link the ubiquitin-proteasome system with the autophagy/lysosome system (Bjorkoy et al. 2005; Kirkin et al. 2009; Lamark et al. 2009; Pankiv et al. 2007), indicating myofilament-dependent cross talk between these two degradation systems in muscles, including the heart (Perera et al. 2010).
MuRF functions for skeletal and cardiac muscles were widely assessed in knockout and transgenic mouse models. Single and double knockouts for MuRF1 (Bodine et al. 2001; Fielitz et al. 2007a; Koyama et al. 2008; Maejima et al. 2014; Wadosky et al. 2014; Willis et al. 2007, 2013; Witt et al. 2008), MuRF2 (Willis et al. 2007, 2013; Witt et al. 2008), and/or MuRF3 (Fielitz et al. 2007a, b), as well as transgenic mouse models for MuRF1 (Wadosky et al. 2014; Willis et al. 2009a), highlighted the importance of these muscle-specific E3-ligases for muscle development and maintenance, as well as for muscle atrophy and hypertrophy.
Sequencing of MuRF1 genes in patients affected with hypertrophic cardiomyopathy led to the discovery of three abnormal variants (Chen et al. 2012). These mutated MuRF1 proteins mis-localized to sarcomeric Z-discs and displayed reduced substrate ubiquitylation, resulting in the pathological accumulation of myosin heavy chain 6, cardiac myosin-binding protein-C, calcineurin, and mTOR in cardiomyocytes.
12.3.2 Cullin-Type E3-Ligases
Cullin-based E3-ligases belong to one of the largest families of E3-ubiquitin ligases. Although the cullin family consists of only eight members (cullin1, cullin2, cullin3, cullin4A, cullin4B, cullin5, cullin7, and cullin9/PARC), their modular assembly poises them for a large number of possible targets (Fig. 12.5). Cullins do not bind their substrate proteins directly, but rely on an array of substrate adaptor protein families. Each of these adaptor protein families contains a specific domain that allows them to interact with a specified set of cullin proteins. Cullin1- and cullin7-based E3-ligases rely on the small protein Skp1 to interact with F-box-domain-containing proteins, while cullin3-based E3-ligases require BTB/POZ-domain-containing substrate adaptors to link to their targets for poly-ubiquitylation (reviewed in Sarikas et al. 2011).
Most cullin proteins (with the exception of cullin7 and cullin9/PARC) contain in their N-terminus three cullin repeats, which moderate the interaction with their substrate adaptor protein families (Table 12.3). A cullin-homology domain, followed by a conserved lysine that is used for neddylation of the protein, is found towards the C-terminus. The C-terminus of most cullins binds to Rbx1/ROC1 (regulator of cullin), which in turn attracts the activated E2-conjugating enzyme (Fig. 12.5). Rbx1 is the protein that carries the RING domain and fulfills thereby the E3-ligase functionality. Therefore, cullin proteins resemble in their role more that of a scaffolding protein, to attract all binding partners into a functioning E3-ligase complex that subsequently (poly-)ubiquitylates a specific cellular substrate protein.
Table 12.3
Select substrate adaptors and substrates for cullin-based E3-ligases
E3-ligase | Select potential and confirmed substrates | Disease link |
|---|---|---|
Cullin1-atrogin | Myosin binding protein-C (Mearini et al. 2009) Calcineurin-A (CnA) (Li et al. 2004) MyoD (Tintignac et al. 2005) IkappaBalpha (Usui et al. 2011) FoxO transcription factors (K63-linkage) (Li et al. 2007a) | Hypertrophy (Li et al. 2007a) Pressure overload (Li et al. 2004) Chronic heart failure (Li et al. 2007b) Phenylephrine and pressure overload-induced hypertrophy, heart failure (Spaich et al. 2012) |
Cullin1-Fbxl22 | Alpha-actinin (Spaich et al. 2012) Filamin-C (Spaich et al. 2012) | |
Cullin1-Fbxo25 | Nkx2.5 (Jang et al. 2011) Isl1 (Jang et al. 2011) Hand1 (Jang et al. 2011) Mef2C (Jang et al. 2011) | |
Cullin1-Skp2 | Cyclin-dependent kinase inhibitor p27 (Chen et al. 2008; Pramod and Shivakumar 2014; Tamamori-Adachi et al. 2004) Akt (Chan et al. 2012) | Ischemia-reperfusion after myocardial infarction (Tamamori-Adachi et al. 2004) Herceptin-induced cardiotoxicity (Chan et al. 2012) |
Cullin1-cdc4/Fbxw7 | Notch 1 and 4 (Tetzlaff et al. 2004) Cyclin E (Tetzlaff et al. 2004) Calcineurin (Kishi et al. 2007) PGC-1alpha (Olson et al. 2008) | Mitochondrial function (Haemmerle et al. 2011) Myocardial infarction (Liepinsh et al. 2013) Diabetic cardiomyopathy (Palomer et al. 2013) |
Cullin1-beta-TrCP | STAT1 (Soond et al. 2008) Skp2 (Wei et al. 2007) cdc4/Fbxw7 (Wei et al. 2007) | |
Cullin2-VHL | Metabolic syndrome, obesity, or diabetes (Lei et al. 2008) | |
Cullin3-KCTD6 | sAnk1.5 (Lange et al. 2012) | |
Cullin3-KLHL40 | LMOD3 (Garg et al. 2014) Nebulin (Garg et al. 2014) | |
Cullin3-KLHL41 | NRAP (Lu et al. 2003) | |
Cullin3-KBTBD13 | Nemaline myopathy (Sambuughin et al. 2010) | |
Cullin3-Keap1 | Nrf2 (Fourquet et al. 2010) | |
Cullin4-ctd2/pcna | ||
Cullin4-DDB2 | Cardiac development (Sakamaki et al. 2012) Ischemia-reperfusion injury and heart failure (Fan et al. 2013) | |
Cullin4-SV5 | STAT (Jackson and Xiong 2009) | |
Cullin5-SOCS | IRS2 (Rui et al. 2002) | Viral myocarditis (Knowlton 2008) Diabetic cardiomyopathy (Palomer et al. 2013) |
Cullin7-Fbxw8 | IRS1 (Xu et al. 2008) |
Studies of knockout mice revealed that cullins are important for cell cycle, apoptosis, tumorigenesis, spermatogenesis, as well as vasculogenesis (Zhou et al. 2013). Cullin proteins are widely expressed in the organism, whereas their substrate adaptor proteins usually display more restricted expression patterns.
12.3.2.1 Regulation
Activation of cullin-based E3-ligases is extremely complex and involves a multitude of proteins and signalosome complexes, among them the COP9 signalosome complex, the small ubiquitin-like modifier nedd8 (box in Fig. 12.1), and CAND1/Tip120. Un-neddylated cullins bind to CAND1/Tip120a (Liu et al. 2002). Tip120b, a muscle-specific isoform of TIP120a, was shown to be regulated by a HECT-type E3-ligase (Aoki et al. 1999; You et al. 2003).
The assembly of the active E3-ligase requires the posttranslational modification of cullins by nedd8 (neddylation) and their homodimerization. Neddylation of cullin releases CAND1 and allows further assembly of the E3-ligase. Besides CAND1, the COP9 signalosome complex was found to be important for the regulation of cullin activity, as it proteolytically removes the nedd8 modification, resulting in an inactivation of the E3-ligase (reviewed in Kato and Yoneda-Kato 2009; Schwechheimer 2004). The importance of this signalosome complex for the heart function can be seen in cardiac-specific knockouts for some of its components. Most notably, ablation of COP9 subunit CSN8 results in dilated cardiomyopathy (Su et al. 2010, 2011, 2013), while silencing of CSN5 was shown to influence l-type calcium channels (Kameda et al. 2006).
12.3.2.2 Cullin1-Atrogin1
Atrogin1/MAFbx/Fbxo32 is a muscle-specific cullin-1 substrate adaptor protein that localizes to the sarcomeric Z-disc (Li et al. 2004). The protein contains an F-box domain that binds to Skp1, a nuclear localization domain, and a PDZ-binding motif in its C-terminus (Gomes et al. 2001). Although atrogin1 localizes to the sarcomere due to its interaction with alpha-actinin, its overexpression did not alter alpha-actinin expression levels, indicating that alpha-actinin may not be a substrate for the atrogin-containing cullin-1 E3-ligase (Li et al. 2004). Confirmed substrates for the E3-ligase activity of cullin1-atrogin1 are a mutated version of myosin-binding protein-C that causes familial hypertrophic cardiomyopathy (Mearini et al. 2009), calcineurin-A (Li et al. 2004), as well as several transcription factors, including members of the Forkhead family of transcription factors (FoxO1, FoxO3a) (Li et al. 2007a), MyoD (Tintignac et al. 2005), and IkappaBalpha (Usui et al. 2011) (Table 12.3). While most of the investigated cullin1-atrogin1 substrates are channeled to the proteasome complex for degradation, the K63-linked poly-ubiquitylation of FoxO transcription factors acts as co-activation signal for their transcriptional activity (Li et al. 2007a) (Fig. 12.5). How the cullin1-atrogin1 E3-ligase differentiates between K48- and K63-linked poly-ubiquitylation of its substrates remains to be further investigated.
12.3.2.3 Other Cullin-1 Substrate Adaptors
Atrogin1 is not the only cullin-1-specific substrate adaptor protein with important roles for the heart. Recently, the cardiac-enriched F-box and leucine-rich repeat protein 22 (Fbxl22) was reported to mediate cullin-1-dependent degradation of alpha-actinin and filamin C (Spaich et al. 2012). Fbxl22 contains an N-terminal F-box domain, followed by a largely unstructured C-terminus. Intriguingly, the protein localizes, like atrogin1, to the Z-disc of the sarcomere, where its two characterized cardiac substrates are located.
Fbxo25, a close homologue of atrogin1 (Gomes et al. 2001), was recently identified as substrate adaptor for several important cardiac transcription factors (Jang et al. 2011). Among other tissues, Fbxo25 is enriched in the fetal heart, and its expression is downregulated during heart development (Jang et al. 2011; Tacchi et al. 2010). The protein displays a nuclear localization (Hagens et al. 2006; Jang et al. 2011) and aggregates in a sub-compartment of the nucleus (Manfiolli et al. 2008). Here, Fbxo25 is thought to specifically ubiquitylate the cardiac transcription factors Nkx2.5, Isl1, Hand1, and Mef2C, which are then routed to the proteasome for degradation (Jang et al. 2011). Hence, developmental downregulation of this cullin1 substrate adaptor may drive cardiac differentiation and development.
Skp2, another F-box domain-containing protein, was also shown to play important functions for cardiomyocyte development and in disease. The cullin1-Skp2 E3-ligase was recently demonstrated to regulate the degradation of cyclin-dependent kinase inhibitor p27, possibly in an ERK2-dependent fashion (Chen et al. 2008; Pramod and Shivakumar 2014; Tamamori-Adachi et al. 2004). Terminally differentiated cardiomyocytes display low levels of Skp2 protein, which is actively ubiquitylated, leading to accumulation of the cullin1-Skp2 substrate p27 and cell-cycle arrest. Overexpression of Skp2 led to p27 degradation and stimulation of CDK2, which stimulated proliferation in D1NLS-/CDK4-transfected cardiomyocytes (Tamamori-Adachi et al. 2004). The same group of authors used the expression of Skp2 in D1NLS/CDK4 virally infected cardiomyocytes to investigate improvement of ischemia-reperfusion injury during myocardial infarction (Tamamori-Adachi et al. 2008).
More recently, a link between Skp2 ubiquitylation of Akt and Herceptin sensitivity was uncovered (Chan et al. 2012), indicating that Skp2 may play a role in Herceptin cardiotoxicity (Fuller et al. 2008).
Knockout mice for cdc4/Fbxw7, another F-box domain-containing protein, display malformation of the heart chamber and die at embryonic day 10.5, suggesting significant roles for cullin1-cdc4/Fbxw7-dependent protein degradation mechanisms during cardiac development (Tetzlaff et al. 2004). Tissues from these mice show accumulation of cyclin-E and Notch proteins 1 and 4, presumably through failed ubiquitylation of the putative substrates by this E3-ligase. The PPARgamma (peroxisome proliferator-activated receptor gamma) co-activator PGC-1alpha was also shown to be ubiquitylated through cullin1-cdc4/Fbxw7 E3-ligase (Olson et al. 2008). Regulation of PPAR through PGC-1alpha was shown to be of importance for the metabolic regulation of mitochondrial function (Haemmerle et al. 2011), during myocardial infarction (Liepinsh et al. 2013) and in diabetic cardiomyopathy (Palomer et al. 2013). Although only demonstrated in yeast, the F-box domain-containing protein cdc4/Fbxw7 was recently shown to promote degradation of calcineurin (Kishi et al. 2007). While the action of the cullin1-cdc4/Fbxw7 E3-ligase on calcineurin awaits confirmation in skeletal or cardiac muscle cells, they open another intriguing way to manipulate calcineurin activity and putatively NFAT signaling (Crabtree and Olson 2002).
The beta-transducin repeat-containing protein 1 (beta-TrCP; also called F-box/WD repeat-containing protein 1A) contains, besides its F-box domain, several WD40 repeats towards the C-terminus of the protein. Several lines of evidence exist that indicate a role for the cullin1-beta-TrCP E3-ligase in the ubiquitylation of beta-catenin, IkappaBalpha (Li et al. 2013; Wei et al. 2007; Winston et al. 1999), and STAT1 (Soond et al. 2008), thereby influencing cardiac signaling pathways. Recent evidence also hints at an involvement of the cullin1-beta-TrCP E3-ligase in the degradation of other cullin1 substrate adaptors, namely, Skp2 and cdc4/Fbxw7 (Wei et al. 2007). Intriguingly, the NAD-dependent deacetylase SIRT1 was shown to regulate beta-TrCP degradation (Woo et al. 2013), indicating regulation of this protein presumably via acetylation and subsequent degradation via E3-ligase mediated poly-ubiquitylation.
12.3.2.4 Cullin-2
Cullin2-based E3-ligases bind to their respective substrate adaptor proteins with the help of the elonginBC heterodimer (Kamura et al. 1998). Substrate adaptor proteins that interact with elonginBC contain a characteristic BC-box motif.
One of the best characterized substrate receptors for cullin2-based E3-ligases is the BC-box domain-containing VHL protein (von Hippel-Lindau tumor suppressor). The cullin2-VHL E3-ligase is known to play important roles for HIF (hypoxia inducible factor) ubiquitylation and degradation (Maxwell et al. 1999; Pozzebon et al. 2013). Degradation of the heterodimeric HIF complex is regulated by oxygen, whereby normal cellular oxygen levels lead to the hydroxylation of the O2-sensitive HIFalpha complex subunit by prolyl-hydroxylases (Semenza 1999). The hydroxylated HIF complex is then recognized by cullin2-VHL as substrate and poly-ubiquitylated for subsequent degradation by the 26S proteasome. Decreased cellular oxygen levels, such as during ischemia, lead to inactivation of prolyl-hydroxylases and stabilization of the HIF complex, which then acts as transcription factor. Analysis of HIF pathway functions for the heart implicated its role in a wide range of diseases, including metabolic syndrome, obesity, or diabetes (Girgis et al. 2012).
A recent publication that investigated VHL functions using adipocyte-specific knockouts found that these mice develop a lethal form of postnatal cardiomegaly within the first week after birth, with dramatically hypertrophied ventricular and septal walls, as well as increased angiogenesis (Girgis et al. 2012). Intriguingly, adipocyte-specific deletion of HIF2alpha but not HIF1alpha rescued the cardiomelagy phenotype completely.
In contrast, mice with cardiac-specific deletion of VHL developed heart failure, malignant transformation of the heart, and premature death by 5 months of age (Lei et al. 2008).
12.3.2.5 Cullin-3
In contrast to cullin1, cullin3-based E3-ligases with importance for cardiac development and function are just starting to be investigated. Cullin3 itself was found to associate with the Z-disc in cardiomyocytes (Lange et al. 2012), like many of its substrate adaptor proteins (Bowlin et al. 2013; du Puy et al. 2012; Gupta et al. 2013). Substrate adaptors for this E3-ligase typically contain a BTB/POZ domain, which interacts with a binding site in the cullin3 protein (Canning et al. 2013) (Fig. 12.5).
Recently, KCTD6 was shown to act as cullin3-based substrate adaptor for the muscle-specific isoform of Ank1, called small ankyrin 1.5 (sAnk1.5) (Lange et al. 2012). KCTD6 localized to the M-band and Z-disc region of the sarcomere. The interaction of KCTD6 with sAnk1.5 was mediated by one of the obscurin-binding domains in the regulatory C-terminus of ankyrin, which is also partially present in non-muscle isoforms of Ank1, indicating regulatory functions for the ubiquitously expressed KCTD6 that extend beyond its role in muscle.
Functions of the cullin3-based substrate adaptor KLHL40/KBTBD5 were lately uncovered to be important for thin filament regulation. KLHL40 mutations cause an autosomal recessive form of nemaline myopathy (Ravenscroft et al. 2013). Although mainly characterized as a skeletal muscle disease, nemaline myopathy has been shown to be associated with cardiomyopathy, depending on disease mechanism and origins (Gatayama et al. 2013; Mir et al. 2011; Nagata et al. 2011; Taglia et al. 2013).
It emerged that cullin3-KLHL40 E3-ligases promote stability of nebulin and leiomidin 3 (LMOD3), thereby increasing thin filament durability in cross-striated muscles (Garg et al. 2014). Although further investigations are needed to uncover the exact mechanism, it is thought that cullin3-KLHL40 decreases the K48-linked poly-ubiquitylation of substrates, thereby promoting their folding and stability. KLHL40 is expressed specifically in heart and skeletal muscle (Bowlin et al. 2013), where it localizes to the sarcomeric I-band and A-band. Knockout mice for KLHL40 display neonatal lethality and show severe disruption of sarcomeres in skeletal muscles only (Garg et al. 2014). Intriguingly, two closely related BTB-domain proteins, KLHL41 and KBTBD13, were also implicated in nemaline myopathy development (Gupta et al. 2013; Sambuughin et al. 2010).
Mutations in KLHL41 (also known as KBTBD10, Krp1, sarcosin) were correlated with nemaline myopathy in several patients, who occasionally also displayed cardiac abnormalities, like ventricular septum defects (VSDs) (Gupta et al. 2013). Functional loss in a zebrafish model showed presence of nemaline bodies and led to diminished motor function and sarcomeric disorganization. KLHL41 is expressed in heart and skeletal muscles and displayed strong association with the I-band and Z-disc of the sarcomere as well as occasionally the M-band (du Puy et al. 2012; Gupta et al. 2013). Here it may associate with the nebulin-related anchoring protein (NRAP), which was recently identified as an interaction partner for KLHL41 (Lu et al. 2003). Knockdown experiments indicated that KLHL41 may also be involved in the lateral fusion of adjacent myofibrils (Greenberg et al. 2008). Mature myofilaments were found to be severely decreased in knockdown cells, which may suggest a similar mechanism to KLHL40-mediated stabilization of myofilaments. Further investigations are needed to better understand the molecular pathway by which this substrate adaptor protein regulates myofibril assembly in cardiomyocytes.
Similar to KLHL40 and KLHL41, mutations of KBTBD13 in patients were found to cause an autosomal-dominant form of nemaline myopathy (Sambuughin et al. 2010). KBTBD13 is another substrate adaptor for cullin3-based E3-ubiquitin ligases (Sambuughin et al. 2012). The protein contains an N-terminal BTB/POZ domain that permits interaction with cullin3 and promotes its homodimerization. KLHL40 harbors in its C-terminus a KELCH domain, which is known to mediate protein–protein interactions, presumably linking up to various substrate proteins. In contrast to KLHL40 and KLHL41, KBTBD13 was found to be largely cytoplasmic and did not colocalize with the myofilaments (Sambuughin et al. 2010).
Another cullin3-based substrate adaptor protein with important cardiac functions is Keap1 (Chauhan et al. 2013). Keap1 is best known for its role as cellular mechanism against oxidative stress through its interaction with the transcription factor Nrf2 (Nfe2l2, nuclear factor (erythroid-derived 2)-like 2) (Nordgren and Wallace 2013). Under normal conditions, Keap1 targets Nrf2 for degradation by the ubiquitin-proteasome system. Oxidation of Keap1 by reactive oxygen species leads to disulfide-bridge formation between two Keap1 molecules, thereby altering its conformation, resulting in the stabilization of Nrf2, which is now able to translocate into the nucleus (Fourquet et al. 2010). This pathway is also important in the acute cellular response to doxorubicin-induced ROS production (Nordgren and Wallace 2013) or ROS production after acute exercise stress (Muthusamy et al. 2011).
Keap1 may also be involved in the clearing of poly-ubiquitylated substrates through the autophagy/lysosome pathway. Intriguingly, Keap1 is able to interact with p62/SQSTM1 and LC3 in a stress-dependent manner (Fan et al. 2010). Indeed, genetic ablation of Keap1 led to accumulation of poly-ubiquitylated aggregates and compromised autophagosome formation through decreased LC3 lipidation. The interaction of Keap1 with both p62 and LC3 may also be the reason for the finding that Keap1 itself, as well as three other BTB-domain-containing proteins, GAN1, ENC1, and KLHL41, get degraded in a cullin3-dependent way, however, independent of the ubiquitin-proteasome machinery (Zhang et al. 2005b).
12.3.2.6 Cullin-4
Similar to cullin3, the functions of cullin4-based E3-ubiquitin ligases for the heart are just emerging. Experiments with zebrafish indicated that cullin4a is responsible for positively modulating cardiac development by upregulating the transcription factor tbx5 (Zhao et al. 2015). Cullin4a morphants were found to display greatly reduced cardiac proliferation and heart looping defects. This effect may be due to nuclear functions of cullin4 family members, cullin4a and cullin4b. Indeed, mammalian cullin4b was shown to associate with the polycomb-repressive complex (PRC2) to mono-ubiquitylate histone-2A (H2A) at K119 (Hu et al. 2012). Further experiments in Xenopus identified FADD (Fas-associated death domain-containing protein), an adaptor protein that transmits apoptotic signals and regulates NfkappaB signaling as interaction partner. Correct regulation of FADD proteins plays important roles during heart development. Embryonic manipulation of FADD protein levels led to induction of apoptosis or changes in cardiac chamber sizes (Sakamaki et al. 2012). Inhibition of FADD has been demonstrated to protect against ischemia-reperfusion injury and in heart failure (Fan et al. 2013).
Substrate adaptors for cullin4-based E3-ligases contain a DWD domain (DDB1-binding WD40) that links them via the DDB1 protein to the cullin scaffold (Jackson and Xiong 2009). Identified substrates for this E3-ligase include p21 [via cullin4-CTD2/PCNA (Jackson and Xiong 2009; Nishitani et al. 2008)]; histones H2A, H3, and H4 [via cullin4-DDB2 (Kapetanaki et al. 2006; Wang et al. 2006)]; or STAT1 through 3 [via cullin4-SV5 (Jackson and Xiong 2009)] (Table 12.3).
12.3.2.7 Cullin-5
Little is known about the biological functions of cullin5-based E3-ligases for cardiac development and function. However, a cullin5 substrate adaptor of the SOCS (suppressor of cytokine signaling) protein family has been linked to the stress-induced degradation of insulin receptor substrate-1 and -2 (IRS1, IRS2) (Hu et al. 2013; Rui et al. 2002). All SOCS proteins have been shown to act as cullin5-based substrate adaptors for the degradation of cytokine-induced signaling intermediates, thereby inhibiting the signaling cascade (Babon et al. 2009). Regulation of cytokine signaling in the heart was shown to be of importance during viral myocarditis (Knowlton 2008) or during diabetic cardiomyopathy (Palomer et al. 2013). Interaction of all SOCS proteins with cullin5 requires the elonginBC heterodimer as a cofactor (Babon et al. 2009).
12.3.2.8 Cullin-7
Similarly to cullin1, E3-ligases that are based on cullin7 require Skp1 as intermediary to bind to their respective substrate adaptors (Sarikas et al. 2011). One identified substrate for cullin7-Fbxw8 E3-ligase is insulin receptor substrate-1 (IRS1) (Scheufele et al. 2013; Xu et al. 2008). Loss of cullin7 in muscles was shown to hyperactivate IRS1, leading to phosphorylation of PI3K/AKT and ERK signaling pathways. This role of cullin7 on IRS1 regulation and in insulin signaling may be of importance during ischemia-reperfusion injury (Nagoshi et al. 2005), or in diabetic cardiomyopathy (Liu et al. 2014), although a clear link needs to be established.
A functional role for cullin7 in myocardial infarction was also shown in transgenic mice that express a mutant version of cullin7 (p193/cul7) (Hassink et al. 2009). The reasoning behind these experiments was that cardiomyocytes expressing this cullin7 mutant permit the reentry or preservation of cell-cycle activity (Nakajima et al. 2004). Indeed, experiments with transgenic p193/cul7 mutant mice reported a modest reduction in scar formation and enhancement of cardiac physiology compared to controls.
12.3.3 Other RING-Type E3-Ligases
12.3.3.1 Parkin
Parkin is a HECT-RING hybrid E3-ubiquitin ligase that has been shown to play important roles for mitochondrial homeostasis. A systematic approach to identify dynamically regulated parkin E3-ligase functions identified hundreds of mitochondrial substrate proteins, including bax, mitofusins 1 and 2, and p62/SQSTM1 (Sarraf et al. 2013) (Table 12.4). Although parkin is best characterized for its role in Parkinson’s disease, recent work established a function for this E3-ligase during myocardial infarction (Kubli et al. 2012) and in aging (Kubli et al. 2013). Parkin knockout mice are viable (Goldberg et al. 2003) and display no baseline cardiac phenotype (Kubli et al. 2012). However, parkin deficiency during stress, such as ischemia-reperfusion, led to defects in mitophagy and the accumulation of damaged mitochondria. Consequently, overexpression of parkin protected cardiomyocytes against hypoxia-mediated cell death (Kubli et al. 2012).
Table 12.4
Select cardiac targets for other RING-type E3-ligases
E3-ligase | Select potential and confirmed substrates | Disease link |
|---|---|---|
Parkin | p62 (Sarraf et al. 2013) bax (Sarraf et al. 2013) | Myocardial infarction (Kubli et al. 2012) Aging (Kubli et al. 2013) |
cIAP1, cIAP2, xIAP | Caspase 7 (Huang et al. 2000) RIP1 (Bertrand et al. 2008) Smac/DIABLO (Hu and Yang 2003) | Protection of cardiac fibroblasts from oxidative damage (Philip and Shivakumar 2013) |
Nrdp1/RNF41 | Ischemia-reperfusion injury (Zhang et al. 2011b) Doxorubicin-induced cardiotoxicity (Zhang et al. 2011a) | |
MDM2, MDM4 | MDM2 (Fang et al. 2000) PDE4D5 (Li et al. 2009) ARC (Foo et al. 2007) | Cardiac fibrosis (Zhang et al. 2014) Dilated cardiomyopathy (Xiong et al. 2007) Pressure overload-induced hypertrophy (Balasubramanian et al. 2006) |
c-Cbl | ErbB1 (Waterman and Yarden 2001) EGFR (Levkowitz et al. 1999) FAK (Rafiq et al. 2011) | Ischemia-reperfusion injury (Rafiq et al. 2014) Tissue injury (Rafiq et al. 2011) Pressure overload-induced hypertrophy (Balasubramanian et al. 2006) Diabetes (Gupte and Mora 2006) |
The mechanism of parkin-induced mitophagy involves the mitochondrial protein kinase PINK1 (PTEN-induced putative kinase 1) and the outer mitochondrial membrane proteins mitofusin 1 and 2 (Chen and Dorn 2013; Sarraf et al. 2013; Tanaka et al. 2010). Parkin binds and promotes K63-linked poly-ubiquitylation of mitofusins in a PINK1-dependent manner (Chen and Dorn 2013; Olzmann and Chin 2008). Poly-ubiquitylation of mitofusin and other outer mitochondrial membrane proteins attracts NIX and p62/SQSTM1, which actively bind to lipidated LC3 to encapsulate mitochondria in the nascent autophagosome (Ding et al. 2010; Narendra et al. 2010) and promote the initiation of mitophagy (Figs. 12.2 and 12.5).
12.3.3.2 IAPs
Inhibitor of apoptosis proteins (cIAP1, cIAP2, xIAP) are RING-domain-containing E3-ligases that ubiquitylate proteins in the apoptosis pathway, including RIP1, Smac/DIABLO (second mitochondria-derived activator of caspases), and several caspases (Bertrand et al. 2008; Hu and Yang 2003; Huang et al. 2000) (Table 12.4). E3-ligases cIAP1 and cIAP2 were shown to constitutively poly-ubiquitylate RIP1 (Bertrand et al. 2008), while xIAP (x-linked IAP) was found to promote poly-ubiquitylation of active caspase 3 (Suzuki et al. 2001) (Fig. 12.5). Intriguingly, cIAP2 may also modulate its activity and promote the mono-ubiquitylation of caspases 3 and 7 (Huang et al. 2000). Although the mechanism remains unclear, the authors of the study suggest that mono-ubiquitylation of caspases may act as an intracellular targeting signal modulating the activity of these proteases (Shih et al. 2000).
E3-ligases of the IAP family have been demonstrated to be important for the heart, mainly due to their function as inhibitors of apoptosis and cell death. In cardiac fibroblasts, cIAP2 was shown to protect the cells from oxidative damage (Philip and Shivakumar 2013). IAP E3-ligases were also found to be upregulated in pressure overload-induced hypertrophy and heart failure (Balasubramanian et al. 2006) and may protect cardiomyocytes in a beta3-integrin-dependent way (Johnston et al. 2009).
12.3.3.3 Nrdp1/RNF41
Nrdp1/RNF41 is a member of the TRIM (tripartite interaction motif) protein family and shows thereby similarities to the domain organization of the MuRF proteins. Most of Nrdp1’s functions have been associated with the ubiquitylation of membrane receptors ErbB2, ErbB3, or ErbB4 of the neuregulin receptor family (Diamonti et al. 2002; Yen et al. 2006) (Table 12.4), indicating that this E3-ligase associates with the cellular membrane. Mechanistically, monomeric Nrdp1 is sufficient to ubiquitylate neuregulin receptors through the actions of its RING domain and the unique protein C-terminus. However, newer research indicated that this E3-ligase may also oligomerize through its N-terminal BBox domain, which leads to auto-ubiquitylation and lability of the protein, indicating a putative mechanism for its regulation (Printsev et al. 2014). Cardiac expression of the neuregulin receptors (like ErbB2) has been shown to be important for cardiovascular development, and their targeted deletion or inhibition through the anti-cancer drug Herceptin (monoclonal antibody against ErbB2) results in either embryonic lethality or cardiotoxic effects (Fuller et al. 2008). Therefore, it is not surprising that cardiac overexpression of the E3-ligase, which in turn regulates ubiquitylation of neuregulin receptors, exacerbates ischemia-reperfusion-induced cardiac injury (Zhang et al. 2011b) through downregulation of ErbB3 or doxorubicin-induced cardiac dysfunction (Zhang et al. 2011a).
12.3.3.4 MDM2, MDM4/MDMX
The MDM2 (murine-double minute) E3-ligase is best known for its role in the degradation of the proapoptotic transcription factor p53 (Honda et al. 1997; Toth et al. 2006) (Table 12.4). Its function to poly-ubiquitylate p53 is enhanced by the closely related MDM4/MDMX (Pei et al. 2012). Both E3-ligases harbor the substrate-binding site (e.g., for p53) in the N-terminal part of the protein, while the C-terminus contains the RING domain (Cheok et al. 2010). Depending on protein levels, MDM2 is able to mono- or poly-ubiquitylate p53 (Li et al. 2003). Poly-ubiquitylation of p53 leads to its nuclear export, where it is degraded by the 26S proteasome (Haupt et al. 1997; Pei et al. 2012; Schuster et al. 2007). In addition to p53 poly-ubiquitylation, MDM2 was also demonstrated to be able to undergo auto-ubiquitylation and to ubiquitylate MDM4/MDMX (Chao 2015). Substitution of the RING domain in MDM2 with that of an unrelated E3-ligase of the RING-type family did not affect the ability of MDM2 to promote auto-ubiquitylation. However, the chimeric MDM2 protein was unable to ubiquitylate p53, suggesting some level of specificity of the RING domain towards p53 (Fang et al. 2000).
Global and cardiac-specific knockout mice for MDM2 exhibit early embryonic lethality due to abnormally increased p53 levels, which can be rescued by genetic deletion of p53 (Grier et al. 2006; Jones et al. 1995; Montes de Oca Luna et al. 1995). Inducible global p53 knockout mice became moribund within 1–2 days and displayed drastic cardiac fibrosis after interrupted tamoxifen treatment (Zhang et al. 2014). While embryonic deletion of MDM2 in a cardiomyocyte-specific MDM2 knockout mouse model displayed no obvious cardiac differences at embryonic stage E9, all knockout mice died at embryonic stage E13.5 and displayed severe morphological defects of the heart (Grier et al. 2006). Endothelial/endocardial-specific knockout of MDM2 also resulted in early embryonic lethality, displaying severe vascular and morphogenic defects (Zhang et al. 2012). Overexpression of MDM2 on the other hand promoted cardiomyocyte survival, blocked cardiac hypertrophy, and protected against ischemia-reperfusion injury in a cell-culture model (Toth et al. 2006).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


