Fig. 8.1
Schematic overview of heart development in higher vertebrates. The early heart tube has a primitive phenotype (light purple). Chamber myocardium (gray) expands rapidly, whereas primary myocardium (purple) of the sinus horns (sh), atrioventricular canal (avc), primary ring (pr), and outflow tract (oft) retain a more primitive phenotype. Abbreviations: a atrium, avn atrioventricular node, avb atrioventricular bundle, lv left ventricle, rv right ventricle, la left atrium, ra right atrium, san sinoatrial node, avj atrioventricular junction, lbb left bundle branch, rbb right bundle branch, pvcs peripheral ventricular conduction system, scv superior caval vein, ift inflow tract, ev embryonic ventricle
Although the SAN is the dominant pacemaker, the other components of the CCS also have intrinsic pacemaker activity. Pacemaker activity or automaticity is the capacity to generate an action potential autonomously. The SAN is dominant because its firing rate is the highest compared to the other components, which are stimulated before they will initiate an action potential autonomously. Because of this so-called overdrive suppression, these components are latent pacemakers that can take over pacing in case of dysfunction of the SAN or AV block [1].
8.2 Development of the Cardiac Conduction System
Early in development, during folding of the embryo, two cardiac mesodermal regions fuse and form a heart tube. At this stage, the heart tube contracts slowly in a peristaltic pattern from the inflow tract towards the cranially located outflow tract. All cardiomyocytes are able to depolarize spontaneously; however, already at this stage, dominant pacemaker activity resides at the inflow tract of the heart. The phenotype of the primary myocardium of the embryonic heart resembles that of the nodal tissues in displaying automaticity a poorly developed contraction apparatus and slow conduction of the electrical impulse [2–4].
Lineage analyses have revealed that the initial heart tube only contributes to the LV and the AVC [5–7]. All other structures within the heart are formed by a continuous process of addition of cells at the inflow and outflow tract and dorsal mesocardium until this structure is ruptured [5, 8]. During the process of looping, a working myocardial gene program is activated at specific sites, resulting in an increase in the proliferation rate and the expression of genes associated with the early chamber myocardium, including natriuretic peptide A (Nppa) and connexin 40 (Cx40). These specific regions will give rise to the future atria and ventricles by rapid expansion (Fig. 8.1). Conduction in these structures is fast [9, 10]. However, they are separated by flanking myocardial cells that retain the primitive slow-conducting properties, the sinus venosus (SV), atrioventricular canal (AVC) inner curvatures, and outflow tract (OFT) [2–4]. The dominant pacemaker activity still resides in the SV, which implies that the newly added cells at the inflow tract immediately acquire this property [11]. The AVC now functions to slow the impulse before it reaches the ventricles, thus ensuring the atria and ventricles to contract in a subsequent pattern. The ECG derived from this configuration of alternating slow- and fast-conducting components resembles that of the adult heart, implying that conduction system function has been established before its morphologically distinguishable parts have been formed [12].
The SAN forms within the SV myocardium; the embryonic AVC myocardium forms the definitive AV node, AV junction (rings), and a large part of the base of the LV. The primary myocardium of the OFT differentiates to ventricular myocardium and forms the RV (and smaller LV) outflow tract. At the border between the LV and RV, an interventricular ring of primary myocardium is present (Fig. 8.1). Here, the interventricular septum will be formed containing the future AVB at its crest. The BBs arise from subendocardial cardiomyocytes in the septum, whereas the PVCS will form from the embryonic trabeculae. The origin and known signaling pathways, transcription factors, ion channels, and gap junctions involved in the development and functioning of the components of the CCS will be addressed in the following paragraphs and are summarized in Fig. 8.2.
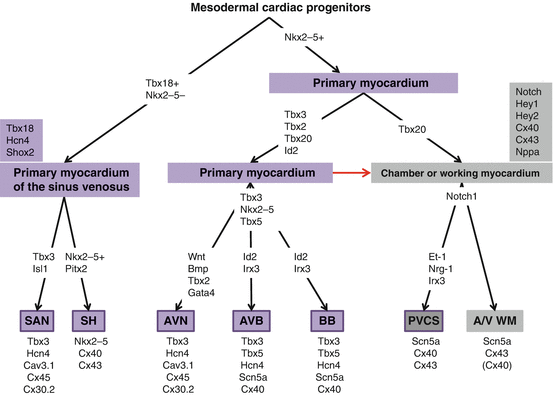
Fig. 8.2
Model for cardiac progenitor and cardiomyocyte differentiation into the myocardial components of the heart and the proteins involved in the regulation and/or correct functioning. The red arrow indicates continuous differentiation of primary cardiomyocytes into chamber myocardium. Nkx2-5, Gata4, and Tbx5 have a broad role in many processes of heart development. They are only included in the scheme when a specific role in CCS formation has been experimentally determined
8.3 Development of the Sinus Venosus and the Sinoatrial Node
The SAN is located at the superior venous entrance of the right atrium. It comprises a few thousand cardiomyocytes forming a “comma-shaped” elongated structure with a head and a tail [13, 14]. The periphery contains connective tissue which protects the SAN from the hyperpolarized right atrium and which dictates the electrical connections with the atrium [15]. SAN cardiomyocytes are glycogen rich and have poorly developed myofibrils and low intercellular conductance properties, which are the result of the expression of the connexin proteins Cx45, Cx30.2, and Cx30, subunits for low conductance gap junctions [16, 17]. Cx40 and Cx43 subunits for high conductance gap junctions are expressed in the atria but not in the SAN [18–21]. Furthermore, the essential cardiac voltage gated sodium channel Nav1.5 (encoded by SCN5A) is expressed at low levels in the SAN, whereas the hyperpolarization activated cyclic nucleotide-gated potassium channel 4 (Hcn4), a key player in the pacemaker potential, is expressed in the mature SAN and absent from the atria [1, 20, 22, 23].
The SV myocardium upstream of the right atrium comprises the SAN, the venous side of the venous valves, and both the right and left sinus horns. The latter will form the sinus coronaries in human. The myocardium of the SV is formed from mouse E9-9.5 onwards from T-box transcription factor (Tbx) 18 and Hcn4-expressing and NK2 homeobox 5 transcription factor (Nkx2-5)-negative precursors. The SV myocardium initially maintains this profile, does not express atrial working myocardial genes such as Cx40, and harbors dominant pacemaker activity. As soon as the SV is formed, a Tbx3-positive domain is observed in the right-sided part, which likely represents the primordium of the SAN. During the fetal period, Hcn4 expression becomes restricted to the Tbx3-positive SAN, whereas the remainder of the SV acquires a working myocardial phenotype [24, 25]. Within the same period, dominant pacemaker activity becomes confined to the actual SAN structure.
Genetic lineage tracing using the Cre-Lox system revealed that the SV and SAN originate from Tbx18-expressing mesenchymal precursors that do not express Nkx2-5, which marks the first and second heart field precursors. Closer inspection of the spatiotemporal expression patterns suggested that Nkx2-5 was briefly expressed in the precursors but downregulated before differentiation [26–29]. Tbx18-deficient mice do not properly form the SV and fail to form the head of the SAN [27].
T-box transcription factor Tbx3 is expressed in the SAN and other components of the CCS, except for the PVCS, and is required to suppress atrial working myocardial genes (Cx40, Cx43, Scn5a) in the SAN [20, 30]. Tbx3 confers the pacemaker phenotype to cardiomyocytes, as ectopic expression in the atria results in the expression of pacemaker genes (e.g., Hcn4) and spontaneous activity in the atria [20]. Mice with hypomorphic alleles of Tbx3 showed bradycardia and sinus pauses indicative of SAN dysfunction [31]. In Nkx2-5-deficient embryos, Tbx3 and Hcn4 are ectopically expressed in the heart tube, indicating that Nkx2-5 suppresses the SV/SAN program in the heart tube [24]. Short stature homeobox 2 gene (Shox2) is specifically expressed in the SV and represses Nkx2-5. The absence of Shox2 results in a hypoplastic SV and upregulation of Nkx2-5 and downregulation of Hcn4 and Tbx3 in the SAN primordium. Consistent with the loss of the pacemaker program, Shox2-deficient mice show bradycardia [32, 33]. Analysis of Shox2-deficient hearts identified LIM homeodomain transcription factor Islet1 (Isl1) as a downstream target. In a zebrafish model, it was shown that Shox2-deficiency-caused bradycardia which was rescued by islet1 (Isl1) overexpression [34]. Isl1 marks the pacemaker cells of the zebrafish heart and is required for its development [35]. Furthermore, lowering Tbx5 expression by a hypomorphic or knockout allele results in a reduction of Shox2 and Tbx3, showing that Tbx5 acts upstream of them [36, 37]. Interestedly, a slight reduction in Tbx5 levels, in mice heterozygous for a hypomorphic Tbx5 allele, results in twofold reduction in Shox2 and Tbx3 expression, suggesting that they are very sensitive to Tbx5 dosage [36]. The right sidedness of the SAN is controlled by the transcription factor paired-like homeodomain 2 (Pitx2), expressed in the left side of the atria and SV. Deficiency in Pitxc2 results in two indistinguishable SANs at both sides of the sinoatrial junction, suggesting that this factor is necessary to suppress SAN formation in the left SV [38].
8.4 Development of the Atrioventricular Node and Atrioventricular Junction
The AVJ includes the AVN, a complex and heterogeneous structure consisting of different cell types with distinct gene expression profiles. The AVJ also encompasses the left and right AV ring bundles that contact anteriorly at the “retroartic node,” thus forming a ring [1, 30, 39, 40]. The AVN and AVJ cells are more primitive than working cardiomyocytes. Their expression profile reflects their pacemaking and slow-conducting properties. The mouse AVN and AVJ express Cx45, Cx30.2, Cav3.1, Hcn4, and Tbx3, whereas Cx40, Cx43, and Scn5a are virtually absent. The anatomical position, expression profile, and slow-conducting property of the AVJ resemble that of the embryonic AVC, suggesting that the AVJ is derived from the AVC [41, 42]. The AVC, first visible around E9-9.5, retains a primitive phenotype when the working myocardial gene program is activated in the future atria and ventricles. In contrast to the fast-conducting working myocardium, the AVC conducts slowly, resembling the function of the AVN in the formed heart [4].
Retrospective clonal analyses revealed a common origin of AV conduction system cells with adjacent working myocardial cardiomyocytes, revealing a cardiogenic origin of the avian CCS [43, 44]. Genetic lineage analysis of Tbx2-positive AVC myocardium revealed that the embryonic AVC forms the AVN, left and right AV ring bundle, and a large part of the free wall of the LV, but not the AVB and BBs [7]. The neural crest does not contribute to the myocardial part of the AVN cardiomyocytes [45].
Tbx3 and Tbx2 are expressed within the AVC and act as repressors with overlapping functions [30, 46, 47]. Tbx2 expression in the heart is lost during the late fetal period, whereas Tbx3 expression is maintained. The retention of the primitive phenotype in the AVC largely depends on the repressive function of Tbx2 and Tbx3. Both proteins compete with Tbx5 for binding of T-box elements in enhancers of T-box target genes (e.g., Nppa and Cx40) and interaction with Nkx2-5 [48]. The loss of both proteins results in loss of the AVC phenotype shown by the expansion of the chamber program (Nppa and Cx40) expression in this region and failure to form AV cushions [47]. Tbx3-deficient embryos have no AVC abnormalities but do show defects in the Tbx2-negative SAN and AVB [49]. Postnatally, correct function of the AVN is dependent on Tbx3 in a dose-dependent manner. Lowering or deleting Tbx3 causes AV conduction abnormalities [31].
The excreted bone morphogenetic protein (Bmp) 2 is expressed specifically in the embryonic AVC and required for AVC specification through activation of the bone morphogenetic protein receptor type IA (Bmpr1A/Alk3) and downstream Smad effectors. Inactivation of Bmp2 in the mouse embryo resulted in loss of Tbx2 expression and failure to form the AVC [50]. In chicken, Bmp2 is sufficient to activate expression of Tbx2 and Tbx3 [46, 50]. Abrogation of BMP signaling by inactivation of the type IA receptor Bmpr1a/Alk3 in the AVC results in inadequate formation of the annulus fibrosus and causes preexcitation [51, 52].
Wnt signaling is fundamental to correct AVC formation. Wnt signaling can be subdivided in canonical (via β-catenin) and noncanonical signaling pathways. A study in zebrafish demonstrated that the canonical signaling pathway is necessary and sufficient to induce Bmp4 and Tbx2b expression in the AVC [53].
The transcription factors Tbx5, Gata4, and Nkx2-5 are expressed broadly in the heart and required for chamber formation. Mutations in TBX5 and NKX2-5 cause congenital heart defects and AV conduction defects [54–56]. Mice haploinsufficient for Nkx2-5 have a hypoplastic AVN, AVB, and BBs. In Tbx5 mutant embryos, the expression of Tbx3 and Cx30.2 are reduced. Postnatally, maturation of the AVN is hampered [57–59]. Gata4 heterozygous embryos show a reduction in Cx30.2 expression. In adult mice, Gata4 haploinsufficiency results in shortened PR intervals [56].
Notch signaling and downstream transcription factors Hey (hes-related family bHLH transcription factor with YRPW motif) 1 and Hey2 sharpen the boundary between AVC and the chambers. In chicken, Notch2 activates Hey1 and Hey2 in the chambers, which in turn repress Bmp2 and Tbx2. In the AVC, Tbx2 represses Hey1 and Hey2, thereby constituting a feedback loop that delimits the AVC [60, 61]. Tbx20-deficiency leads to ectopic expression of Tbx2 in the entire heart tube [62, 63]. Tbx20 represses Tbx2 expression in the chambers by interfering with Bmp-signaling-mediated activation of Tbx2 through interacting with Smad1/5 [62, 63].
The annulus fibrosus and central fibrous body electrically insulate the atria from the ventricles. The only myocardial electrical connection between the atria/AVN and ventricles is the AVB. The annulus fibrosus forms by the invasion of subepicardial mesenchyme of the AV groove in-between the atrial/AVJ myocardium and ventricular myocardium [42]. This latter process is initiated in the early fetal period and continues postnatally [64]. The presence of ectopic fast-conducting myocardial connections between the atria and ventricles involves abnormalities in the invasion of the connective tissue and the normally slow-conducting AVC myocardium acquiring fast-conducting properties [65, 66].
Several genes have been implicated in the formation of accessory pathways and ventricular preexcitation. Mutations in PRKAG2 encoding the protein kinase, AMP-activated, and gamma 2 non-catalytic subunit have been associated with ventricular preexcitation due to disruption of the annulus fibrosus [67]. In addition, a 20p12.3 microdeletion that includes BMP2 was found to predispose to Wolff-Parkinson-White syndrome [68]. In mice, the loss of Bmp2 receptor Alk3 causes additional myocardial strands. Inactivation of Tbx2 in the developing heart results in a Cx40-, Cx43-, and Scn5a-positive and presumably fast-conducting accessory pathway on the left dorsal side of the AVC [65]. Activation of Notch signaling by overexpression of the Notch1 intracellular domain in the heart results in accessory pathway formation and preexcitation [69]. Altogether, these data reveal a Bmp-signaling-Tbx2-Notch pathway that is involved in transcriptional control of correct development of the AVC and annulus fibrosus [51, 52, 65, 68].
8.5 Origin and Development of the Ventricular Conduction System
The ventricular conduction system (VCS) comprises the AVB, left and right BB, and PVCS. Cardiomyocytes in these structures have a poorly developed contractile apparatus and T-tubular system. Gap junctions in the adult VCS are well developed and consist of Cx40 and Cx43 subunits resulting in fast conduction. Gap junction subunit Cx40 is a specific marker for the complete VCS [70, 71]. As soon as the LV and RV emerge in the heart tube (E9-9.5), a G1N2/Tbx3-positive “primary ring” [30, 72] is present in-between the LV and RV. As the interventricular septum is being formed at this location, its crest differentiates into the future AVB [73]. The BBs are gradually formed subendorcardially during growth of the interventricular septum. Tbx3 is expressed in the BBs in a diminishing gradient towards the apex [49]. Cx43, Tbx18, and Tbx20 are also expressed in the IVS but absent from the Tbx3-positive developing AVB and BBs and are therefore useful as negative markers of the AVB and BBs. Tbx3-deficiency results in ectopic expression of these genes, but also of Nppa and Cx40, within the crest of the septum. This again shows that Tbx3 is required for specification by repressing the ventricular working myocardial phenotype. Although Cx40 initially is suppressed in the AVB by Tbx3, this gene is upregulated after E12 to become highly expressed in the AVB [49].
Tbx5 and Nkx2-5 are expressed in the AVB and BBs and were shown to be important for its correct patterning and specification. Mice haploinsufficient for Tbx5 and Nkx2-5 do not establish an AVB and develop conduction block after birth. This defect is caused partly by the failure to activate inhibitor of differentiation protein 2 (Id2) in the AVB [57]. Furthermore, Tbx5 is required to activate both Cx40 and Scn5a in the developing and mature AVB [74]. We speculate that the relative level of Tbx5 to Tbx3 increases in the AVB, leading to the activation of Cx40. In Tbx3-deficient embryos, inhibitor of DNA binding 2 (Id2) expression is not affected showing that activation of the Id2 locus is independent of Tbx3 and suggesting the presence of independent pathways in the formation of the AV bundle and BBs [49]. Iroquois homeobox 3 (Irx3) expression confines to the AVB, BB, and PVCS during development. Irx3 knockout mice display prolonged ventricular activation due to asynchronous activation of the left and right ventricles. Irx3 represses Cx43 and indirectly activates the expression of Cx40 in the AVB and BB [75].
The embryonic ventricular chambers mainly consist of trabecules and a thin outer layer. The impulse preferentially conducts over the trabecules [76]. Early on, the LV is entirely Cx40-positive, whereas the RV is partially Cx40-positive [71], a pattern reflecting that of its essential activator Tbx5 [77]. With further development, the compact walls form (>E11), and expression of Cx40 becomes confined to the trabecular zone. After birth, Cx40 expression is confined further to the subendocardial VCS. This pattern suggests that the Cx40-positive embryonic trabecular myocardium gives rise to both the Cx40-positive VCS and the Cx40-negative compact wall [41]. Prospective and retrospective genetic labeling experiments substantiated the hypothesis derived from the Cx40 (and Nppa) expression pattern that the embryonic trabecules give rise to the VCS and that the compact wall myocardium lineage gradually diverges from the initially Cx40-positive embryonic ventricular walls [71].
In the chick, endothelin 1 (Et1) is an inductive signal secreted from the endocardium and coronary arterial endothelium. Et1 and its receptors were found to be involved in the functional maturation of the VCS and in the induction of expression Cx40 (in chick a subendocardial and periarterial VCS is present) [78]. In mice, neuregulin-1 (Nrg-1) influences patterning of the PVCS, probably through its role in trabecular development [79]. In addition, Notch1 mutants display defective trabeculation mediated by reduced expression of Nrg-1 and Bmp10 [80]. Bmp10 is known to be important for cardiomyocyte proliferation [81]. Recently, it was shown that Notch signaling plays an important role in specification of prenatal ventricular cardiomyocytes towards a conduction phenotype that resembles the PVCS [82].
Conclusion
Our basic understanding of the development and correct functioning of the CCS has been greatly enhanced over the recent years. Genetic lineage tracing uncovered the progenitor pools of the heart and the CCS and a multitude of factors organized in transcriptional circuits have been identified [83, 84]. Genome-wide analyses of tissue-specific transcription factor-DNA interactions (ChIP-seq) and transcriptional profiling (RNA-seq) reveal how these circuits orchestrate gene expression patterns within the heart. Regulatory DNA sequences have been discovered and the 3D conformation of genomic loci unraveled [85]. Recently, the role of histone modifications, chromatin remodeling complexes, and other epigenetic factors has been implicated in CCS development [86, 87]. Together, these data provide new insights and a better understanding into the mechanisms underlying the establishment and maintenance of the CCS.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


