Cardiac Biochemistry
The biochemistry of cardiac tissue involves tightly regulated interactions among ions, proteins, receptors, second messenger systems, and various cellular structures as well as extracardiac influences. Several abnormalities involving neurohormonal pathways as well as derangements of the contractile apparatus of the cardiac myocyte have been demonstrated in cells isolated from failing hearts. This chapter reviews some of the salient features of the biochemistry of the cardiac myocyte.
CARDIAC CONTRACTILITY AND CALCIUM HOMEOSTASIS
The cardiac myocyte is an interconnected network of myofibrils surrounded by sarcoplasmic reticulum (SR). Each myofibril comprises sarcomeres made up of thick myosin filaments and thin actin filaments that form the basic contractile unit of the cardiac myocyte (Fig. 6.1). The active sites of the actin filaments are covered in the resting state by two regulatory proteins, tropomyosin and troponin. Intracellular Ca2+ is the most important determinant of myocardial contractility and relaxation.1 Once contraction ensues, calcium (Ca2+) entry through L-type Ca2+ channels triggers an exponential release of Ca2+ from the SR through ryanodine receptors.2 Calcium then binds troponin, leading to a conformational alteration of tropomyosin, exposing the actin active site, facilitating a “sliding” interaction between the actin filaments and the myosin heads as well as the hydrolysis of ATP (adenosine triphosphate), thus providing energy for contraction.3 Following a cycle of excitation–contraction coupling, diastolic relaxation is initiated by cytosolic Ca2+ sequestration in the SR by the SR-Ca2+ ATPase (SERCA2a) pump (~75%) and exportation extracellularly by the Na+/Ca2+ exchanger (~25%) located on the sarcolemmal membrane.4,5 Abnormal cardiac SR function and Ca2+ signaling represent a characteristic of both systolic and diastolic Congestive Heart Failure (CHF).6–8 There is good evidence that changes in either expression or function of specific calcium-handling proteins lead to increased intracellular diastolic Ca2+ levels, decreased intracellular Ca2+ transients, delayed Ca2+ efflux, and depressed contractility.9,10,11,26 For example, ryanodine receptors are upregulated and progressively activated by phosphorylation, contributing to the SR Ca2+ leak observed in CHF. Phospholamban is a regulatory protein that exerts an inhibitory effect on SERCA2a, limiting its ability to remove cytosolic Ca2+ following contraction. In failing hearts, phospholamban expression is altered and SERCA activity is decreased, resulting in diastolic and systolic dysfunction.5,6,12,13,14–17 An understanding of the molecular mechanisms of heart failure is necessary to understand the potential for therapeutic targets. This fact is underscored by the recent clinical trial investigating whether the function of SERCA can be restored via gene transfer.18
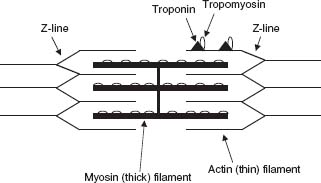
FIGURE 6.1 Sarcomere anatomy.
β-ADRENERGIC SIGNALING
Three β-adrenergic receptor (β-AR) subtypes have been characterized, β1, β2, and β3. Catecholamines act to increase myocardial contractility primarily through β1-adrenergic receptor stimulation leading to G protein–mediated adenyl cyclase activation and cyclic adenosine 3′5′ monophosphate (cAMP) generation, which triggers protein kinase A (PKA)–dependent phosphorylation of voltage-gated L-type Ca+ channels, ryanodine receptors, and phospholamban, which derepresses SERCA2a, leading to excitation–contraction coupling and positive inotropy (Fig. 6.2).19–21 β2-Adrenergic receptors as well as muscarinic cholinergic receptors, through an inhibitory G protein, provide negative control of adrenergic stimulation by inactivating adenyl cyclase, thereby limiting the generation of cAMP.22 The role of β3-adrenergic receptors is poorly defined, but there is some evidence that β3-adrenergic receptors maintain coronary vasomotor tone through the nitric oxide (NO) pathway.23 β-Arrestins also serve to restrict cAMP generation by increasing cAMP degradation and desensitizing the β-receptor.24 Derangements in chronic β-adrenergic signaling that have been implicated in the pathogenesis of CHF include β-AR downregulation, β-AR uncoupling from second messenger systems, upregulation of β-adrenoreceptor kinase (βARK1), and altered calcium trafficking.25–30 β-Receptor blockade can restore calcium homeostasis and upregulate SERCA2a, ultimately improving cardiac performance with long-term treatment.31
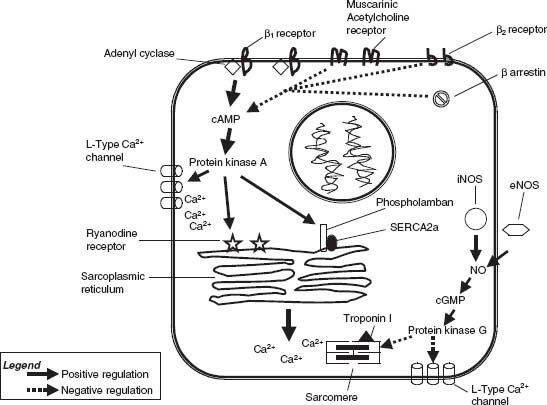
FIGURE 6.2 β-Adrenergic and NO regulation of the cardiac myocyte.
DIGITALIS AND THE NA+-K+ ATPASE
Digitalis, a cardiac glycoside derived from the foxglove plant, has been used for centuries to treat heart failure and atrial fibrillation. Digitalis functions by inhibiting the sodium pump (Na+/K+ ATPase) found in the cardiac cell membrane.32,33 The Na+/K+ ATPase works constitutively, using energy from the hydrolysis of ATP to maintain a high intracellular K+ concentration and a high extracellular Na+ concentration.34 Ca2+ is removed from the cytosol into the extracellular fluid by a sodium–calcium exchange (NCX1) pump driven by the preexisting Na+ gradient.35 Inhibiting the Na+/K+ ATPase promotes enhanced Na+/Ca2+ exchange, leading ultimately to increased intracellular Ca2+ being available to the contractile apparatus, potentially leading to increased myocardial contractility.
PHOSPHODIESTERASE INHIBITION
Phosphodiesterase inhibitors (PDIs) such as milrinone affect contractility by inhibiting phosphodiesterase 3 (PDE3), increasing intracellular cAMP and Ca2+, which leads to increased inotropy.36 PDIs also have vasodilating properties that are important in unloading the failing ventricle.37 Unfortunately, the gain in cardiac performance is tempered by increased arrhythmogenesis, myocardial oxygen consumption, and cardiac death, mitigating its usefulness beyond being a bridge to cardiac transplantation in end-stage CHE.38,39
RENIN–ANGIOTENSIN SYSTEM
The renin–angiotensin system (RAS) has a detrimental role in the pathogenesis of heart failure. Beyond its influences on blood pressure and salt and water regulation, it has stimulatory effects on the sympathetic nervous system, direct effects on myocardial hypertrophy, and indirect effects on myocardial contractility. Numerous large randomized clinical trials have demonstrated the symptom relief and survival benefit in patients with CHF treated with angiotensin-converting enzyme (ACE) inhibitors.40–42 Angiotensinogen is cleaved to angiotensin I by the renally produced enzyme renin in response to renal hypoperfusion. Angiotensin I is then cleaved by ACE into the potent vasoconstrictor angiotensin II. Angiotensin II stimulates catecholamine release, increases cardiac hypertrophy, regulates blood pressure (angiotensin II receptors), and increases blood volume by stimulating aldosterone and vasopressin release, enhancing sodium and water retention (Fig. 6.3).43 ACE inhibitors also increase the generation of bradykinin (thought to mediate the cough associated with ACE inhibitors), which is a nitric oxide synthase (NOS) agonist and may attenuate β-adrenergic contractility through NO signaling.44 Bradykinin degradation may also have untoward effects on myocardial contractility that are offset by ACE inhibition.45
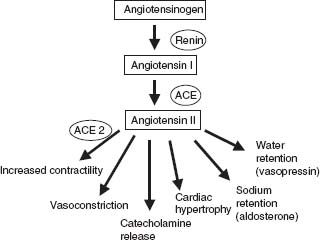
FIGURE 6.3 Renin–angiotensin system.
ACE-2 is an ACE isoform that is thought to be an important regulator of cardiac contractility. It catalyzes the cleavage of angiotensin I to angiotensin 1–9 and of angiotensin II to angiotensin 1–7. ACE-2 is not inhibited by ACE inhibitors, nor is bradykinin a by-product of its activity.46–48 ACE-2 is upregulated within the myocardium with angiotensin II receptor blockade.49 ACE-2 deficiency diminishes cardiac contractility and upregulates hypoxia-induced genes, suggesting its role in RAS modulation following ischemiamediated cardiac injury.50
NITRIC OXIDE
NO plays an important role in the endothelium-dependent functions of coronary vasomotor tone and thrombogenesis, but it also has direct effects on myocardial relaxation. NO is generated by the enzyme NOS, which has three isoforms: eNOS (endothelial), iNOS (inducible), and nNOS (neuronal). NO affects myocardial relaxation through effects on excitation–contraction coupling, regulation of adrenergic signaling, and mitochondrial metabolism.51 Attenuation of β-adrenergic stimulation by NO (see Fig. 6.2) is mediated by cyclic guanosine 3′5′-monophosphate (cGMP)–dependent phosphodiesterase EII regulation of cAMP levels, protein kinase G–mediated downregulation of L-type Ca+ channels,52,53 and the desensitization of troponin I to calcium.54 NO may also influence myocardial relaxation by enhancing the activity of the delayed rectifier K+ current55 as well as cGMP-mediated inhibition of phospholamban and its negative control over SERCA2a.56
REFERENCES
1. Morgan JP. Abnormal intracellular modulation of calcium as a major cause of cardiac contractile dysfunction. N Engl J Med. 1991;325:625.
2. Lehnart SE, Wehrens XH, Kushnir A, et al. Cardiac ryanodine receptor function and regulation in heart disease. Ann N Y Acad Sci. 2004;1015:144–159.
3. Guyton AC. Textbook of Medical Physiology. 8th ed. Philadelphia: WB Saunders; 1991:68–72.
4. Schulze DH, Muqhal M, Lederer WJ, et al. Sodium/calcium exchanger (NCX1) macromolecular complex. J Biol Chem. 2003;278(31):28849–28855.
5. Arai M, Matsui H, Periasamy M. Sarcoplasmic reticulum gene expression in cardiac hypertrophy and heart failure. Circ Res. 1994;74:555–564.
6. Schmidt U, Hajjar RJ, Helm PA, et al. Contribution of abnormal sarcoplasmic reticulum ATPase activity to systolic and diastolic dysfunction in human heart failure. J Mol Cell Cardiol. 1998;30:1929–1937.
7. SE, Maier LS, Hasenfuss G. Abnormalities of calcium metabolism and myocardial contractility depression in the failing heart. Heart Fail Rev. 2009;14(4):213–224.
8. Gwathmey JK, Copelas L, Mackinnon R, et al. Abnormal intracellular calcium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76.
9. Whitmer JT, Kumar P, Solaro RJ. Calcium transport properties of cardiac sarcoplasmic reticulum from cardiomyopathic Syrian hamsters (BIO 53.58 and 14.6): evidence for a quantitative defect in dilated myopathic hearts not evident in hypertrophic hearts. Circ Res. 1988;62:81–85.
10. Gwathmey JK, Slawsky MT, Hajjar RJ, et al. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J Clin Invest. 1990;85:1599–1613.
11. Hasenfuss G, Mulieri LA, Leavitt BJ, et al. Alterations of contractile function and excitation contraction coupling in dilated cardiomyopathy. Circ Res. 1992;70:1225–1232.
12. Schmidt AG, Zhai J, Carr AN, et al. Structural and functional implications of the phospholamban hinge domain: impaired SR Ca2+ uptake as a primary cause of heart failure. Cardiovasc Res. 2002;56(2):248–259.
13. Mercadier JJ, Lompre AM, Duc P, et al. Altered sarcoplasmic reticulum Ca2(+)-ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Invest. 1990;85: 305–309.
14. Kadambi VJ, Ponniah S, Harrer JM, et al. Cardiac-specific overexpression of phospholamban alters calcium kinetics and resultant cardiomyocyte mechanics in transgenic mice. J Clin Invest. 1996;97:533–539.
15. Hajjar RJ, Schmidt U, Kang JX, et al. Adenoviral gene transfer of phospholamban in isolated rat cardiomyocytes. Rescue effects by concomitant gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase. Circ Res. 1997;81:145–153.
16. Hajjar RJ, Schmidt U, Matsui T, et al. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci U S A. 1998;95:5251–5256.
17. Ogletree ML, Sweet WE, Talerico C, et al. Duration of left ventricular assist device support: Effects on abnormal calcium cycling and functional recovery in the failing human heart. J Heart Lung Transplant. 2010;29(5):554–561.
18. Hajjar RJ, Zsebo K, Deckelbaum L, et al. Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail. 2008;14:355–367.
19. van der Heyden MA, Wijnhoven TJ, Opthof T. Molecular aspects of adrenergic modulation of cardiac L-type Ca2+ channels. Cardiovasc Res. 2005;65(1):28–39.
20. Gao MH, Ping P, Post S, et al. Increased expression of adenylyl cyclase type VI increases β-adrenergic receptor-stimulated production of cAMP in neonatal rat myocytes. Proc Natl Acad Sci U S A. 1998;95:1038–1043.
21. Freeman K, Lerman I, Kranias EG, et al. Alterations in cardiac adrenergic signaling and calcium cycling differentially affect the progression of cardiomyopathy. J Clin Invest. 2001;107:967.
22. Dorn GW, Molkentin JD. Manipulating cardiac contractility in heart failure: data from mice and men. Circulation. 2004;109:150–158.
23. Dessy C, Moniotte S, Ghisdal P, et al. Endothelial beta3–adrenoceptors mediate vasorelaxation of human coronary microarteries through nitric oxide and endothelium-dependent hyperpolarization. Circulation. 2004;110(8):948–954.
24. Perry SJ, Baillie GS, Kohout TA, et al. Targeting of cyclic AMP degradation to beta 2-adrenergic receptors by beta-arrestins. Science. 2002;298(5594):834–836.
25. Bristow MR, Minobe W, Rasmussen R, et al. Beta-adrenergic neuroeffector abnormalities in the failing human heart are produced by local rather than systemic mechanisms. J Clin Invest. 1992;89:803–815.
26. Bristow MR, Ginsburg R, Minobe W, et al. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211.
27. Akhter SA, Skaer CA, Kypson AP, et al. Restoration of betaadrenergic signaling in failing cardiac ventricular myocytes via adenoviral-mediated gene transfer. Proc Natl Acad Sci U S A. 1997; 94:12100–12105.
28. Rockman HA, Chien KR, Choi DJ, et al. Expression of a betaadrenergic receptor kinase inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A. 1998;95:700–705.
29. Harding VB, Jones, LR, Lefkowitz RJ. Cardiac βARK1 inhibition prolongs survival and augments β-blocker therapy in a mouse model of severe heart failure. Proc Natl Acad Sci U S A. 2001;98:5809.
30. Engelhardt S, Hein L, Dyachenkow V, et al. Altered calcium handling is critically involved in the cardiotoxic effects of chronic beta-adrenergic stimulation. Circulation. 2004:109(9):1154–1160.
31. Plank DM, Yatani A, Ritsu H, et al. Calcium dynamics in the failing heart: restoration by beta-adrenergic receptor blockade. Am J Physiol Heart Circ Physiol. 2003;285(1):H305–H315.
32. McDonough AA, Velotta JB, Schwinger RH, et al. The cardiac sodium pump: structure and function. Basic Res Cardiol. 2002; 97(suppl 1):I19–I24.
33. Gheorghiade M, Adams KF, Colucci WS. Digoxin in the management of cardiovascular disorders. Circulation. 2004; 109:2959–2964.
34. Smith TW, Antman EM, Friedman PL, et al. Digitalis glycosides: mechanisms and manifestations of toxicity: part I. Prog Cardiovasc Dis. 1984;26:495–530.
35. Hilgemann DW. New insights into the molecular and cellular workings of the cardiac Na+/Ca2+ exchanger. Am J Physiol Cell Physiol. 2004;287(5):C1167–C1172.
36. Yano M, Kohno M, Ohkusa T, et al. Effect of milrinone on left ventricular relaxation and Ca(2+) uptake function of cardiac sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. 2000;279(4):H1898–H1905.
37. Sonnenblick EH, Grose R, Strain J, et al. Effects of milrinone on left ventricular performance and myocardial contractility in patients with severe heart failure. Circulation. 1986;73(3 pt 2): III162–III167.
38. Chatterjee K, De Marco T. Role of nonglycoside inotropic agents: indications, ethics, and limitations. Med Clin North Am. 2003;87(2):391–418.
39. Cuffe MS, Califf RM, Adams JF Jr, et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA. 2002;287(12):1541–1547.
40. Pfeffer MA, Braunwald E, Moye LA, et al. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N Engl J Med. 1992;327(10):669–677.
41. The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325(5):293–302.
42. The CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N Engl J Med. 1987;316(23):1429–1435.
43. Paul M, Pinto YM, Schunkert H, et al. Activation of the reninangiotensin system in heart failure and hypertrophy: studies in human hearts and transgenic rats. Eur Heart J. 1994;15:63–67.
44. Wittstein IS, Kass DA, Pak PH, et al. Cardiac nitric oxide production due to angiotensin-converting enzyme inhibition decreases beta-adrenergic myocardial contractility in patients with dilated cardiomyopathy. J Am Coll Cardiol. 2001;38(2):429–435.
45. Krombach RS, McElmuray JH III, Gay DM, et al. Bradykinin degradation and relation to myocyte contractility. J Cardiovasc Pharmacol Ther. 2000;5(4):291–299.
46. Boehm M, Nabel EG. Angiotensin-converting enzyme 2—a new cardiac regulator. N Engl J Med. 2002;347(22):1795–1797.
47. Crackower MA, Sarao R, Oudit GY, et al. Angiotensinconverting enzyme-2 is an essential regulator of heart function. Nature. 2002;417(6891):822–828.
48. Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1–9. Circ Res. 2000;87(5):E1–E9.
49. Ishiyama Y, Gallagher PE, Averill DB, et al. Upregulation of angiotensin-converting enzyme 2 after myocardial infarction by blockade of angiotensin II receptors. Hypertension. 2004;43(5):970–976.
50. Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26(4):369–375.
51. Massion PB, Feron O, Dessy C, et al. Nitric oxide and cardiac function: ten years after, and continuing. Circ Res. 2003;93:388–398.
52. Han X, Kobzik I, Balligand JL, et al. Nitric oxide synthase (NOS3)-mediated cholinergic modulation of Ca2+ current in adult rabbit atrioventricular nodal cells. Circ Res. 1996;78:998–1008.
53. Gallo MP, Ghigo D, Bosia A, et al. Modulation of guinea-pig cardiac L-type calcium current by nitric oxide synthase inhibitors. J Physiol. 1998;506(pt 3):639–651.
54. Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457–467.
55. Bai CX, Namekata I, Kurokawa J, et al. Role of nitric oxide in Ca2+ sensitivity of the slowly activating delayed rectifier K+ current in cardiac myocytes. Circ Res. 2005;96(1):64–72.
56. Zhang Q, Scholz PM, He Y, et al. Cyclic GMP signaling and regulation of SERCA activity during cardiac myocyte contraction. Cell Calcium. 2005;37(3):259–266.



