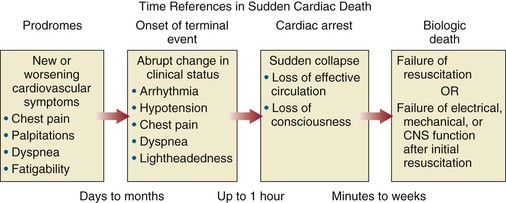
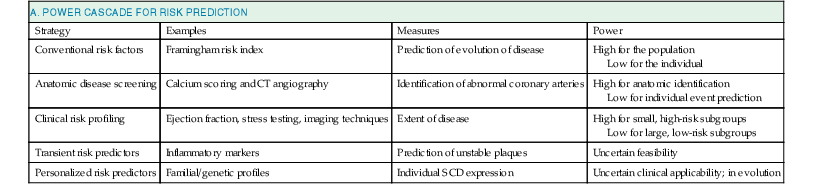
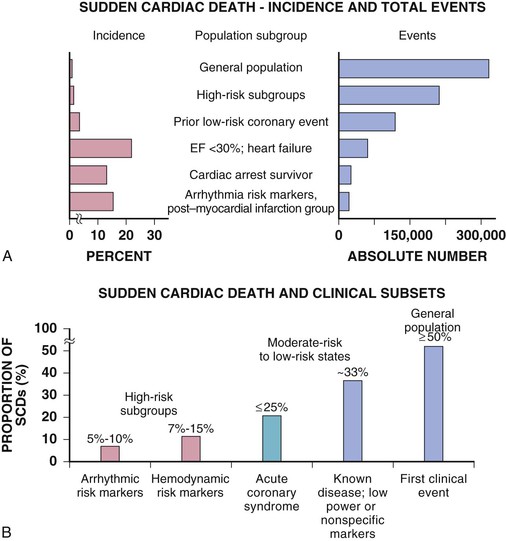
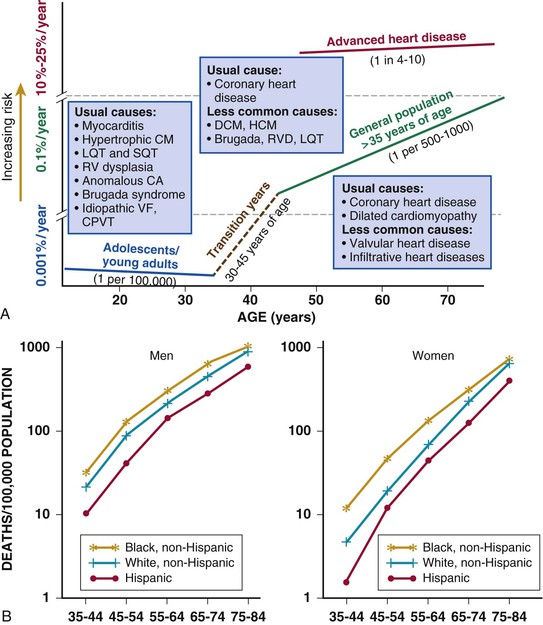
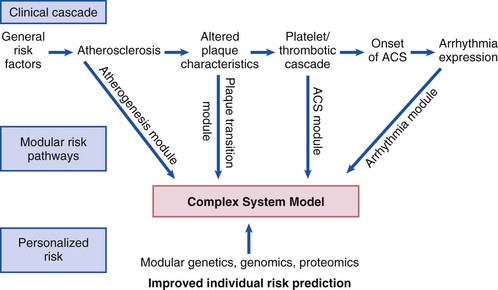
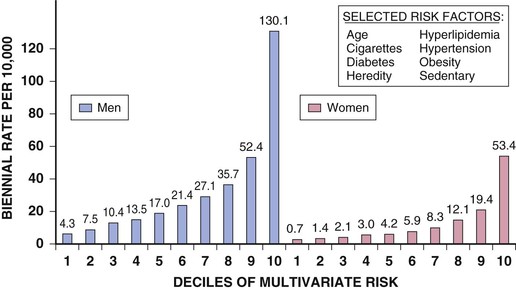
Robert J. Myerburg, Augustin Castellanos Sudden cardiac death (SCD) is a major public health problem because of its frequency and demographics. With numeric estimates in the range of 300,000 to 375,000 deaths per year in the United States alone, it accounts for half of all cardiovascular deaths. Approximately 50% of all SCDs are unexpected first expressions of a cardiac disorder, often striking during the victim’s productive years. Despite recognition of an association between forewarning symptoms of chest pain or syncope and SCD dating to Hippocrates around 400 bc, the description of a “shrunken and withered” artery to the heart in a victim of SCD in the late 1490s by Da Vinci, and an epidemiologic survey in Rome by Lancisi at the request of Pope Clement XI in1706, advances in prediction, prevention, and management of unexpected cardiac arrest and SCD did not begin to emerge until 50 years ago. It is anticipated that the major insights into causes, pathophysiology, and preventive and management strategies developed during the past few decades, described in this chapter, will continue to evolve. SCD is natural death from cardiac causes heralded by abrupt loss of consciousness within 1 hour of the onset of an acute change in cardiovascular status (Table 39-1). Preexisting heart disease may or may not have been known to be present, but the time and mode of death are unexpected. This definition incorporates the key elements of natural, rapid, and unexpected. It consolidates previous definitions that have conflicted, mainly because the most useful operational definition of SCD in the past differed for clinicians, cardiovascular epidemiologists, pathologists, and scientists attempting to define pathophysiologic mechanisms. As the epidemiology, clinical expression, causes, and mechanisms began to be understood, these differences merged. To satisfy clinical, scientific, legal, and social considerations, four temporal elements must be considered: (1) prodromes, (2) onset, (3) cardiac arrest, and (4) biologic death (Fig. 39-1). Because the proximate cause of SCD is an abrupt disturbance in cardiovascular function followed by loss of consciousness, any definition must recognize the brief time interval between onset of the mechanism directly responsible for cardiac arrest and the consequent loss of blood flow. Therefore the 1-hour definition, which primarily refers to the duration of the “terminal event,” defines the interval between the onset of symptoms signaling the pathophysiologic disturbance leading to cardiac arrest and the onset of the cardiac arrest itself. Prodromes, occurring weeks or months before an event, are not sensitive or specific predictors of an impending event, but premonitory signs and symptoms, which can occur during the days or weeks before cardiac arrest, may be more specific for imminent cardiac arrest when they begin abruptly. Sudden onset of chest pain, dyspnea, or palpitations and other symptoms of arrhythmias often precede the onset of cardiac arrest and define the 1-hour onset of the terminal event that brackets the cardiac arrest. The fourth element, biologic death, was an immediate consequence of cardiac arrest in the past and usually occurred within minutes. However, the generally accepted clinical-pathophysiologic definition of up to 1 hour between onset of the terminal event and biologic death requires qualifications for specific circumstances. For example, since the development of community-based interventions and life support systems, patients may now remain biologically alive for a long period after the onset of a pathophysiologic process that has caused irreversible damage and will ultimately lead to death. In this circumstance, the causative pathophysiologic and clinical event is the cardiac arrest itself rather than the factors responsible for the delayed biologic death. Thus death remains defined biologically, legally, and literally as an absolute and irreversible event timed to cessation of all biologic functions, but most studies link the definition of SCD to the cardiac arrest rather than to a biologic death that occurs during hospitalization after cardiac arrest or within 30 days. Finally, forensic pathologists studying unwitnessed deaths continue to use the definition of sudden death for a person known to be alive and functioning normally 24 hours before, and this remains appropriate within obvious limits. Epidemiologic studies of SCD are difficult to interpret for both theoretical and practical reasons. There are persisting inconsistencies about the definition and challenges in accessing data and adjudicating individual cases in data sets, in determining pathophysiologic mechanisms, and in making distinctions between population risk and individual risk.1 In addition, the fact that sudden cardiac arrest (SCA) leading to SCD has short-term dynamics superimposed on a long-term static or dynamic substrate introduces unusual epidemiologic complexities, including long-term risk prediction based on the evolution of atherogenesis, myocardial hypertrophy, and ventricular muscle dysfunction over time and modulation by transient (short-term) variables such as ischemia, hemodynamic shifts, atherosclerotic plaque disruption and thrombosis, and autonomic variations. The differences between chronic disease evolution and transient events call for different forms of epidemiologic modeling (Table 39-2A). Furthermore, the emerging field of genetic epidemiology adds another dimension for study, and there is a need for focusing on interventional epidemiology, a term coined to define the population dynamics of therapeutic outcomes. In reference to risk for SCD from coronary heart disease, clinical categories ranging from general population risk to personalized risk profiling are paralleled by the partition of risk predictors into the pathophysiologic categories of substrate-based risk and expression-based risk2 (see Table 39-2B). Substrate-based risk refers to prediction of the evolution or identification of vascular or myocardial substrates that establish risk for SCD (i.e., atherogenesis, scar patterns, remodeling) and to quantification of these risks. It should not be perceived as limited to anatomic features because molecular variants may also provide risk substrates. In contrast, expression-based risk refers to the identification of mechanisms and pathways that contribute to the clinical manifestation of the risk established by the substrate. This category includes plaque transition and acute coronary syndromes (plaque disruption and thrombogenesis) and their potential for specific expression as an arrhythmic event in susceptible individuals. The arrhythmogenic category of risk can also be viewed to include modifiers of molecular-based risk that drive individual expression. The worldwide incidence of SCD is difficult to estimate because numbers vary as a function of the prevalence of coronary heart disease in different countries (see Chapter 1).3 The annual number of SCDs in the United States is derived from multiple sources, such as retrospective death certificate data, American Heart Association (AHA) statistical updates based on data from the National Center for Health Statistics,4 and national extrapolations from a large emergency rescue experience in one community5 and a community-wide multisource data set from another.6 Recently, data from large surveillance studies, such as the ROC (Resuscitation Outcomes Consortium), are contributing additional insight into the subtleties of data collection and interpretation.7 Three factors are of primary importance for identification of populations at risk and consideration of strategies for prevention of SCD: (1) the absolute numbers and event rates (incidence) among population subgroups (Fig. 39-2A), (2) the clinical subgroups in which SCDs occur (Fig. 39-2B), and (3) the time dependence of risk (Fig. 39-3). When the more than 300,000 adult SCDs that occur annually in the United States are viewed as a global figure for an unselected adult population 35 years and older, the overall incidence is calculated to be in the range of 0.1% to 0.2% per year (1 to 2/1000 population; see Fig. 39-2A). This general population includes the large proportion of SCDs that occur as a first clinical manifestation, as well as SCDs that can be predicted with greater accuracy in higher risk subgroups (see Fig. 39-2B). Any intervention designed for the general population must therefore be applied to the 999 per 1000 who do not have an event to reach and possibly influence the 1 per 1000 who does, in contrast to the much smaller subsets that can be profiled at higher risk. Figure 39-2A highlights this problem by expressing the incidence (percent per year) of SCD among various subgroups and comparing the incidence figures with the total number of events that occur annually in each subgroup. Thus, despite the large absolute number at risk in the general population and the impact of proven interventions on populations, the practical ability of applying the principles of population risk to targeted individual patients is challenging. The cost and risk-to-benefit uncertainties limit the nature of such broad-based interventions and demand a higher resolution of risk identification. Two fundamental approaches for attacking this challenge can be followed: a general population strategy targeting prevention of acquired risk factors such as obesity (primordial prevention) and primary prevention by control of manifest risk factors14 and a more focused individual risk strategy based on identification and intervention in small subsets of the general population with a high density of risk (Fig. 39-4). On moving from the total adult population to a subgroup at higher risk because of the presence of selected coronary risk factors, there may be a 10-fold or greater increase in the annual incidence of events, with the magnitude being dependent on the number and types of risk factors operating in specific subgroups. The size of the denominator pool, however, remains very large, and implementation of interventions remains problematic, even at this heightened level of risk. Higher resolution is desirable and can be achieved by identification of more specific subgroups. However, the corresponding absolute number of deaths becomes progressively smaller as the subgroups become more focused (see Fig. 39-2A), thus limiting the potential benefit of interventions to a much smaller fraction of the total number of patients at risk. Up to half of all SCDs attributable to coronary heart disease are first clinical events,2 and another 20% to 30% occur in subgroups of patients with known coronary heart disease who are profiled to be at relatively low risk for SCD on the basis of current clinically available markers (see Fig. 39-2B). The principle of a high proportion of SCDs occurring as first events or in previously asymptomatic individuals applies to the less common causes as well.15 Temporal elements in risk for SCD have been analyzed in the context of both biologic and clinical chronology. In the former, epidemiologic analyses of risk for SCD in populations have identified three patterns: diurnal, weekly, and seasonal. General patterns of heightened risk during the morning hours, on Mondays, and during the winter months have been described.16 An exception to the diurnal risk pattern is SCD in sleep apnea, in which the risk tends to be nocturnal.17 Ambient temperature is an environmental factor associated with risk for SCD. Both excessive cold18 and excessive heat19 have been linked to risk for cardiac arrest, although the studies did not determine whether temperature extremes are associated with ventricular tachyarrhythmias versus other mechanisms of cardiac arrest. Another environmental variable, transient ambient air pollution conditions, has been correlated with an increased incidence of ventricular arrhythmias stored in implantable cardioverter-defibrillator (ICD) memories,20 but the question of whether these are cardiac arrest equivalents is uncertain. In the longer-term clinical paradigm, risk for SCD is not linear as a function of time after changes in cardiovascular status.10,11,21 Survival curves after major cardiovascular events, which identify risk for both sudden and total cardiac death, usually demonstrate that the most rapid rate of attrition occurs during the first 6 to 18 months after an index event (see Fig. 39-3). Thus there is a time dependence of risk that focuses the potential opportunity for maximum efficacy of an intervention during the early period after a conditioning event. Curves that have these characteristics have been generated from among survivors of out-of-hospital cardiac arrest, new onset of heart failure, and unstable angina and from patients with recent myocardial infarction and low ejection fractions or heart failure. For the latter, however, early nonarrhythmic deaths also contribute a large proportion of the fatal events. Even though the rate of attrition decreases after the early spike in mortality, a secondary delayed increase in risk occurs in post–myocardial infarction patients 2 to 5 years after an index event, probably related to ventricular remodeling and heart failure. Age, Race, Sex, and Heredity The incidence of sudden death has two peak ages: within the first year of life (including sudden infant death syndrome [SIDS]; see Chapter 62) and between 45 and 75 years of age. Among the general populations of infants younger than 1 year and middle-aged or older adults, the incidence is surprisingly similar.22 In adults older than 35 years, the incidence of SCD is 1 per 1000 persons per year (Fig. 39-5A), with an age-related increase in risk over time as the prevalence of coronary heart disease increases as a function of advancing age.10 The incidence in infants is 73 per 100,000 person-years, and the incidence in adolescents and adults younger than 30 years is approximately 6 per 100,000 person-years,22,23 or 1% of the risk in middle-aged and older adults (Fig. 39-5A). In contrast to incidence, however, the proportion of deaths caused by coronary heart diseases that are sudden and unexpected decreases with advancing age. In the 20- to 39-year age group, approximately 75% of the deaths attributable to coronary heart disease in men are sudden and unexpected, with the proportion falling to approximately 60% in the 45- to 54-year age group and hovering close to 50% thereafter. Age also influences the proportion of any cardiovascular cause among all causes of natural sudden death in that the proportion of coronary deaths and of all cardiac causes of death that are sudden is highest in the younger age groups whereas the fraction of total sudden natural deaths that result from any cardiovascular cause is higher in the older age groups. At the other end of the age range, only 19% of sudden natural deaths in children between 1 and 13 years of age have cardiac causes; the proportion increases to 30% in the 14- to 21-year age group. In the transition age range between adolescence and young adulthood (to the age of 25 years) and in the middle and older ages (beginning at 35 years of age), coronary heart disease emerges to its position as the dominant cause of SCD. However, rare disorders, such as hypertrophic cardiomyopathy, Brugada syndrome, long-QT syndrome, and right ventricular dysplasia, are significant contributors to the distribution of causes of SCD in this age group. In one study, myocardial fibrosis of unknown etiology was a significant cause in this age group.24 Race. A number of studies comparing racial differences in the relative risk for SCD in whites and blacks with coronary heart disease in the United States had yielded conflicting and inconclusive data. However, the most recent studies demonstrate a higher risk for cardiac arrest and SCD in blacks than in whites (see Fig. 39-5B and Chapter 2).25 SCD rates in Hispanic populations were lower. These differences were observed across all age groups. Sex. SCD syndrome has a large preponderance in men relative to women during the young adult and early middle-age years because of the protection that women enjoy from coronary atherosclerosis before menopause (see Fig. 39-5B). Various population studies have demonstrated a fourfold to sevenfold greater incidence of SCD in men than in women before 65 years of age, at which point the difference decreases to 2:1 or less, and continues to decrease with advancing age. As risk for coronary events increases in postmenopausal women, risk for SCD increases proportionately, with similar rates in men and women. Even though the overall risk for SCD is much lower in younger women, coronary artery disease is the most common cause of SCD in women older than 40 years, and the classic coronary risk factors, including cigarette smoking, diabetes, use of oral contraceptives, and hyperlipidemia, all influence risk in women (see Chapter 77).26 Data from the Nurses’ Health Study suggest that a healthy lifestyle, defined as no cigarette smoking, a low body mass index, regular exercise, and a healthy diet, reduces the risk for SCD in women by as much as 46% to more than 90%, depending on the number of low-risk markers present.27 Women are 50% less likely to have severe left ventricular dysfunction and 66% less likely to have known coronary heart disease before SCD28 and are therefore less likely to be profiled as high risk and more likely to have SCD as a first cardiac event. Heredity. Familial patterns of risk for SCD, which result from known or suspected genetic variations, are emerging as important factors for risk profiling. This concept is generally applicable in regard to both disease development and SCD expression in the common acquired disorders and in a specific sense to inherited arrhythmogenic conditions associated with SCD. The various genetic associations can be separated into four categories (Table 39-3): inherited uncommon primary arrhythmic syndromes (e.g., long-QT syndromes, Brugada syndrome, catecholaminergic polymorphic ventricular tachycardia or fibrillation), inherited uncommon structural diseases associated with risk for SCD (e.g., hypertrophic cardiomyopathy, right ventricular dysplasia), “acquired” or induced risk for arrhythmias (e.g., drug-induced long QT interval or proarrhythmia, electrolyte disturbances), and common acquired diseases associated with risk for SCD (e.g., coronary heart disease, nonischemic cardiomyopathies) (see Chapters 32 and 33). Genetic variants mapped to loci on many chromosomes are being defined as the molecular bases for these entities and associations. The multiple specific mutations at gene loci–encoding ion channel proteins associated with the various inherited arrhythmia syndromes (see Chapter 32) represent a major advance in the understanding of a genetic and pathophysiologic basis for these causes of sudden death. In addition, the role of modifier genes and mutation specificity in the severity of clinical phenotypes in long–QT interval syndromes29,30 and structural diseases such as hypertrophic cardiomyopathy31 is of increasing interest. These observations may provide screening tools for individuals at risk, as well as the potential to devise specific therapeutic strategies. In addition, gene loci identified by genome-wide association studies may also serve as candidates for investigation of the role of low-penetrance mutations or polymorphisms in SCD caused by more common conditions, such as coronary heart disease.32 At this time it appears that the hope for common variants linked to common syndromes such as SCD will be superseded by multiple rare variant associations. To the extent that SCD is an expression of underlying coronary heart disease, hereditary factors that contribute to risk for coronary heart disease operate nonspecifically for the SCD syndrome. However, studies have identified mutations and relevant polymorphisms along multiple steps of the cascade, from atherogenesis to plaque destabilization, thrombosis, and arrhythmogenesis, each of which is associated with increased risk for a coronary event (Fig. 39-6).33,34 Integration of these individual markers may provide more powerful individual risk prediction in the future. In addition, several studies have suggested that SCD as the initial expression of coronary heart disease demonstrates familial clustering,35–38 including general population surveillance studies, family histories of cardiac arrest survivors in the community, studies of ventricular fibrillation (VF) during acute myocardial infarction, and postmortem evaluation of SCD cases (Table 39-4). TABLE 39-4 Family History of and Risk for Primary Sudden Cardiac Death AMI = acute myocardial infarction; ASHD = arteriosclerotic heart disease; CI = confidence interval; EMS = emergency medical service; Hx = history; MI = myocardial infarction; OR = odds ratio; PCA = primary cardiac arrest; RR = relative risk; STEMI = ST-segment elevation myocardial infarction. Risk profiling for coronary artery disease, by means of the conventional risk factors for coronary atherogenesis, is useful to identify levels of population risk and individual risk but cannot be used to distinguish individual patients at risk for SCD from those at risk for other manifestations of coronary heart disease (see Chapters 49 to 54). Multivariate analyses of selected risk factors (e.g., age, diabetes mellitus, systolic blood pressure, heart rate, electrocardiographic abnormalities, vital capacity, relative weight, cigarette consumption, and serum cholesterol level) have determined that approximately 50% of all SCDs occur in the 10% of the population in the highest risk decile on the basis of multiple risk factors (Fig. 39-7). Thus the cumulative risk derived from multiple risk factors exceeds the simple arithmetic sum of the individual risks. Comparison of risk factors in victims of SCD with those in people in whom any manifestation of coronary artery disease develops does not provide useful patterns, by either univariate or multivariate analysis, to distinguish victims of SCD from the overall pool. However, a history of diabetes mellitus and a tendency to longer QTc intervals on random electrocardiograms are suggested as potential markers of interest for prediction of SCD.39 Although angiographic and hemodynamic patterns discriminate SCD risk from non-SCD risk only under limited conditions, familial clustering of SCD as a specific manifestation of the disease may lead to the identification of specific genetic abnormalities that predispose to SCD.36–38 Hypertension is a clearly established risk factor for coronary heart disease and also emerges as a highly significant risk factor in the incidence of SCD (see Chapters 43 and 44). However, there is no influence of increasing systolic blood pressure levels on the ratio of sudden deaths to total coronary heart disease deaths. No relationship has been observed between cholesterol concentration and the proportion of coronary deaths that were sudden. Neither the electrocardiographic pattern of left ventricular hypertrophy nor nonspecific ST-T wave abnormalities influence the proportion of total coronary deaths that are sudden and unexpected; only intraventricular conduction abnormalities are suggestive of a disproportionate number of SCDs, an old observation reinforced by data from some device trials that suggest the importance of QRS duration as a risk marker. Low vital capacity also suggests a disproportionate risk for sudden versus total coronary deaths. This is of interest because such a relationship was particularly striking in the Framingham Study in analysis of data from women who had died suddenly. The conventional risk factors used in early studies of SCD are risk factors for the evolution of coronary artery disease. The rationale is based on two facts: (1) coronary disease is the structural basis for 80% of SCDs in the United States, and (2) coronary risk factors are easy to identify because they tend to be present continuously over time (see Fig. 39-6). However, risk factors specific for fatal arrhythmias are dynamic pathophysiologic events and occur transiently.40 Transient pathophysiologic events are being modeled epidemiologically in an attempt to express and use them as clinical risk factors for both profiling and intervention.1 Nonetheless, data suggest that longitudinal and transient risk predictors may have their power blunted by clinical interventions, such as percutaneous coronary intervention during acute coronary syndromes and post–myocardial infarction beta blocker therapy.41,42 Identification of specific clinical markers of risk for SCD as a specific expression of both coronary heart disease and other cardiovascular disorders has been a goal for many years.10,11 Left ventricular ejection fraction has been the most popular of such markers for clinical trials and patient profiling. However, its sensitivity limitations and inability to identify the large subgroup in which SCD is the first expression of heart disease have encouraged investigators to seek additional markers. For example, exercise data from a large cohort of men observed for years after a stress test demonstrated that a profile of higher resting heart rates, smaller increments in rate during exercise, and lower decrement in heart rate during the first minute after exercise predicted higher risk for SCD during follow-up.43 In addition, a number of electrocardiographic indicators (such as microvolt T wave alternans and indices of QT duration and dispersion), genetic profiles, and other indices of the extent of disease are also predictive (see Chapters 35 and 37). The Framingham Study demonstrated a striking relationship between functional classification and death during a 2-year follow-up period. However, the proportion of deaths that were sudden did not vary with the functional classification, with a range of 50% to 57% in all groups, including those free of clinical heart disease and those in functional class IV. Other studies have also suggested that patients with heart failure and better functional capacity are at lower risk for dying, as expected, but that a higher proportion of such deaths are sudden.44
Cardiac Arrest and Sudden Cardiac Death
Perspective
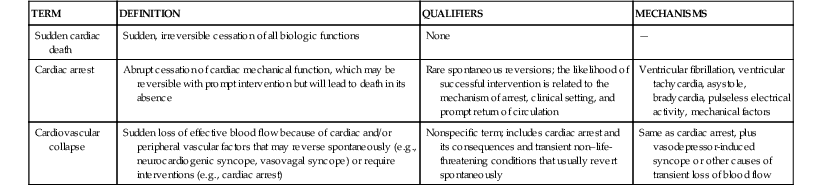
Definitions
Epidemiology
Epidemiologic Overview
Incidence and the Population Burden of Sudden Cardiac Death
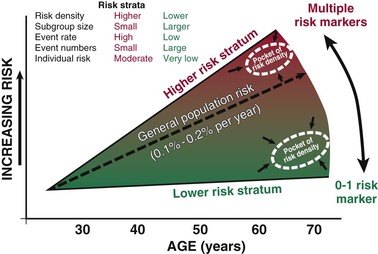
Population Pools, Risk Gradients, and Time Dependence of Risk
Population and Subgroup Risk Versus Individual Risk Assessment
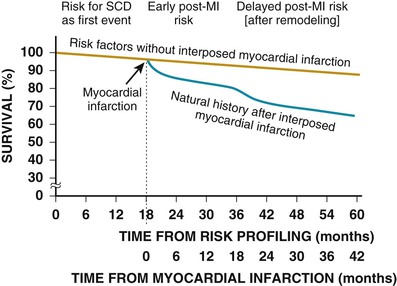
Biologic and Clinical Time-Dependent Risk
STUDY SITE
COHORT
CONTROLS
FAMILY HISTORY MEASURE
OUTCOME
Seattle35
1988-1994
EMS SCA subjects
Population matched
Hx of MI or PCA in 1° relatives
Paris36
1967-1994
Population surveillance
Retrospective analysis
Hx of PCA in 1° relatives
Netherlands37
2001-2005
STEMI with VF
STEMI without VF
Hx of SCD in 1° relatives
Finland38
2000-2003
SCD with AMI
AMI survivors
Population controls
SCD or AMI in 1° relatives without ASHD

Risk Factors for Sudden Cardiac Death
General Profile of Risk for Sudden Cardiac Death (see Chapters 32 and 33)
Functional Capacity and Sudden Death
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cardiac Arrest and Sudden Cardiac Death
39