Fig. 4.1
Excitation–contraction coupling: Ca2+ cycling is regulated by transporters and channels on the plasma membrane and sarcoplasmic reticulum. Under pathological conditions, such as hypertrophy and heart failure, defects in EC coupling manifest as depressed outward K+ currents (particularly I to), L-type Ca2+ current, Na+/Ca2+ exchange, SR Ca2+ uptake due to SERCA2a downregulation, impaired ryanodine receptor function, depressed β-adrenergic receptor signaling, and phospholamban phosphorylation. β-Adrenergic receptor signaling is depressed mostly through a decrease in expression of β-1 receptors. Ca2+-induced Ca2+ release is diminished in part because of reduced sarcoplasmic reticulum Ca2+ content – systolic Ca2+ – stemming from increased RyR2 leak. Diastolic Ca2+ transient is prolonged in part because of reduced expression of SERCA and increased inhibition from phospholamban. A decrease in SR Ca2+ load leads to a decrease in the amount of Ca2+ released through RyR2 and subsequently lower cytosolic Ca2+. Contraction and relaxation are also slowed by shifts in myofilament regulation (e.g., decreased troponin I phosphorylation) and myosin isozyme expression (e.g., increased MHCβ). Abnormalities in myocardial PKC and PKA activities regulate many of these events. Abbreviations: β–1 β-1 adrenergic receptors, Ca 2+ calcium, Gq G-protein subunit q, GαS/Gi G-protein subunits αS/i, K + potassium, Na 2+ sodium, PLCβ phospholipase C subunit beta, cAMP cyclic adenosine monophosphate, PKA protein kinase A, PKC protein kinase C, DAG diacylglycerol, P phosphate, AC adenylyl cyclase, PDE phosphodiesterase, ATP adenosine triphosphate, NE norepinephrine, Ang II angiotensin II, ET–1 endothelin 1, SERCA2a sarcoplasmic/endoplasmic reticulum Ca2+ ATPase
Excitation leads to the opening of voltage-gated L-type Ca2+ channels, allowing the entry of a small amount of Ca2+ into the cell [5]. Through a coupling mechanism between the L-type Ca2+ channel (LTCC) and the SR Ca2+ release channel (ryanodine receptor 2 – RyR2), a larger amount of Ca2+ is released through a process termed Ca2+-induced Ca2+ release (CICR), activating the myofilaments, leading to contraction [5]. This process increases intracellular Ca2+ concentration from approximately 10 nM during diastole to about 1 μM during systole. The relatively increased Ca2+ concentration in the T-tubular SR junctions inactivates the LTCC by a Ca2+-dependent inactivation and consequently terminates Ca2+ influx to avoid Ca2+ overload and arrhythmias [5, 6].
During relaxation, Ca2+ is re-accumulated back into the SR by the SR Ca2+-ATPase pump (SERCA2a) and extruded extracellularly primarily by the sarcolemmal Na+/Ca2+ exchanger-1 (NCX1). The plasma membrane Ca2+-ATPase (PMCA) pump and the mitochondrial uniporter may also contribute to this process, albeit minimally [5]. The contribution of each of these mechanisms for lowering cytosolic Ca2+ varies with species and is mostly determined by the abundance and activity of SERCA2a and sarcolemmal NCX1, by [Na+]i and [Ca2+]i, and by the membrane potential [5]. In humans ~75 % of the Ca2+ is removed by SERCA2a and ~25 % by the NCX1 [7]. The Ca2+ pumping activity of SERCA2a is influenced by phospholamban (PLN) [8] and sarcolipin [9]. In the unphosphorylated state, PLN inhibits SERCA2a activity, whereas phosphorylation of PLN by cAMP-dependent protein kinase A (PKA) and by Ca2+/calmodulin-dependent protein kinase (CaMKII) reverses this inhibition [8]. CaMKII can also directly phosphorylate SERCA2a and enhances its activity [10]. Sarcolipin on the other hand inhibits SERCA2a through direct binding and through stabilization of SERCA2a–PLN interaction in the absence of PLN phosphorylation and through the inhibition of PLN phosphorylation [9]. Disease-induced malfunction of several of these key EC coupling proteins and the subsequent alterations in intracellular Ca2+ homeostasis result in mechanical and electrical dysfunction at the molecular level, cellular level, and the entire organ.
Although Ca2+ regulation of EC coupling is fundamental for cardiac physiology, Ca2+ is also essential for the regulation of an array of Ca2+-dependent regulatory processes associated with cardiac remodeling. There is emerging evidence that the induction of this Ca2+-dependent remodeling is not affected by the rapid elevation in the contractile Ca2+ stimulus but is mediated by a more sustained Ca2+ signal. Several reports have now described the existence of spatially restricted and distinct Ca2+ pools in cardiomyocytes that influence contractility and Ca2+-dependent signaling pathways. How the cardiac myocyte distinguishes between contractile Ca2+ and signaling Ca2+ has been a matter of debate and few hypotheses have been put forward to explain this conundrum [3, 4]. However, a picture is now emerging that emphasizes how spatially restricted Ca2+ signals in local Ca2+ nanodomains, microdomains, and macrodomains can selectively recruit certain transcription factors and not others, or activate certain metabolic molecules and not others, through activation of diverse Ca2+-permeable ion channels with widely differing timescales of activation.
A classic example of the concept of Ca2+ microdomains is represented by the maintenance of a very close approximation of LTCC and RyR2 in the dyads between T-tubular sarcolemma and the junctional SR that promotes contraction [11]. A similar microdomain also exists between mitochondria and the SR that facilitates an efficient and rapid mitochondrial Ca2+ uptake to meet the bioenergetic demand of the beating myocyte [12]. Other examples of Ca2+ microdomains involve local Ca2+ pools provided and regulated by (1) LTCC [13], T-type Ca2+ channels (TTCC), and plasma membrane Ca2+-ATPase isoform 4 (PMCA4) in non-junctional sarcolemmal caveolae [14]; (2) transient receptor potential channel (TRPC) proteins [15]; and (3) inositol 1,4,5-triphosphate receptor (IP3R) localized to the nuclear membrane [16], which are all believed to be implicated in mediating diverse Ca2+-responsive signal transduction in hypertrophy and heart failure.
4.2.2 Ca2+-Induced Ca2+ Entry
Although calcium signaling is a combination of a highly organized Ca2+ release from intracellular Ca2+ stores – SR – and Ca2+ entry across the plasma membrane, extracellular Ca2+ influx is necessary to maintain cytosolic Ca2+ levels. In non-excitable cells, the mechanism that controls Ca2+ entry is triggered by intracellular Ca2+ store depletion which activates plasma membrane Ca2+ channels and elicits a prolonged increase in intracellular Ca2+ in a process now termed store-operated Ca2+ entry (SOCE). This mechanism of Ca2+ influx plays a critical role in replenishing Ca2+ stores and providing a sustained and precise local Ca2+ signals that are critical in determining a spectrum of downstream cellular responses.
STIM1 (stromal interacting molecule), a recently identified single-span ER membrane molecule, has a unique and essential role in mediating Ca2+ entry through SOCE [17, 18]. STIM1 contains four low-Ca2+-affinity luminal EF hands which are sensitive to ER Ca2+ concentration; when ER Ca2+ is high, Ca2+ binds to STIM1 EF hands and inactivates it. However, when ER Ca2+ is decreased, Ca2+ dissociates from the EF hands and causes conformational changes that result in the activation of STIM1. So, STIM1 functions as sensor of Ca2+ levels in the lumen of the ER and activator of the SOCE channel(s) in the plasma membrane [19]. STIM1, through its Ca2+-binding EF hands, can detect small changes in ER Ca2+ levels and responds by rapidly oligomerizing and translocating into specialized regions close to the ER–plasma membrane junctions where it interacts with and activates plasma membrane Orai channels [20, 21]. Interaction of STIM1 with Orai Ca2+ channels then triggers entry of Ca2+ and replenishment of intracellular Ca2+ homeostasis and activation of Ca2+-dependent signaling pathways [22]. As Ca2+ levels increase in the ER lumen or at ER–plasma membrane junctions – microdomains – STIM1–Orai coupling is turned off and STIM1-induced Orai channel activation is terminated, leading to STIM1 protein de-oligomerization [23, 24].
Whether cardiomyocytes possess a similar functional SOCE mechanism is not clear since Ca2+ entry through SOCE requires depletion of SR/ER Ca2+ store content, a condition which is hardly achieved in vivo in physiological conditions. However, SOCE has emerged as a potential mechanism regulating Ca2+ transients in cardiomyocytes, and a number of recent studies have provided evidence that SOCE is, for instance, operational in neonatal cardiomyocytes [25, 26]. Hulot et al. have reported that STIM1 is abundant in neonatal cardiomyocytes but marginally expressed in adult cardiomyocytes [26]. Interestingly, these studies also reported that STIM1 manipulation did not have any influence on cardiac EC coupling. These observations were further confirmed by Luo et al. who have also demonstrated that SOCE is nearly absent in adult cardiomyocytes but highly active in neonatal myocytes or adult cardiomyocytes under pathological cardiac remodeling [27]. This latter study also demonstrated that STIM1 expression levels correlate with the magnitude of SOCE activation and the subsequent increase in Ca2+ entry [27].
Although the SOCE process has been initially reported to mainly result from the coupling of STIM1 with Orai Ca2+ channels [25], STIM1 is now known to interact with and activate several transient receptor potential (TRP) channels, which have long been linked to SOCE [28]. Recent evidence suggests, for example, that TRPC1, TRPC3, and TRPC6 are implicated in cardiac hypertrophic responses [29, 30]. Moreover, studies have reported that STIM1 interacts with and controls the function of Cav1.2 voltage-operated Ca2+ channel in smooth muscle and neuron [31, 32]. STIM1 interacts with the C-terminal end of the LTCC and inhibits its function and decreases its surface expression, thus leading to its inactivation [32]. Interestingly, this action of STIM1 on LTCC is thought to favor Ca2+ entry via Orai channel [32]. Given the importance of LTCC in Ca2+ signaling in cardiac hypertrophy, it is not known whether STIM1 has a similar effect on LTCC in cardiomyocytes, an observation that remains to be determined.
Although STIM1 appears to function as a master regulator of SOCE protein complexes, many questions pertaining to its role in cardiomyocytes remain to be answered. For instance, what are the coupling partners of STIM1 in the heart? Do Orai and TRP channels act synergistically or redundantly and do they have different timescales of activation? Do they activate a common Ca2+ pool and/or do they elicit different Ca2+-dependent signaling pathways? What is the role of STIM2, Orai1, Orai2, Orai3, and other TRP channels in the heart? However, emerging results from a number of studies show that adult cardiomyocytes may not need SOCE per se to regulate SR Ca2+ loading or contractile Ca2+ pools, but this process has a functional significance in fetal and neonatal cardiomyocytes and during cardiac growth and remodeling [15].
4.3 Calcium Signaling in Hypertrophy and Heart Failure
4.3.1 Intracellular Ca2+ Handling in Hypertrophy and Heart Failure
The remarkable low cytosolic Ca2+ gradient is tightly maintained, as noted earlier, by the concerted activity of several highly conserved families of Ca2+-handling pumps, channels, and transporters that regulate a balance between Ca2+ entry, extrusion, and storage. Although initial modifications of Ca2+ handling are beneficial, several lines of evidence now suggest that Ca2+ dysregulation contributes to pathological hypertrophy and heart failure (Chaps. 3 and 16).
4.3.1.1 SERCA2a, PLN, and Ca2+ Regulation
Heart failure is characterized by a number of abnormalities at the cellular level in the various steps of EC coupling. The major abnormality in Ca2+ cycling that occurs in heart failure is the observation that SR Ca2+ stores are significantly reduced. When this store is depleted, SR Ca2+ release is curtailed, in terms of both its amplitude and duration, and as a result reduced contraction force is generated. The likely cause of this deficiency (depressed SR load) is the differential changes in gene expression and activity of key Ca2+ regulatory proteins. Ca2+ transients recorded from failing human myocardial cells or trabeculae reveal a significantly prolonged Ca2+ transient with an elevated end-diastolic intracellular Ca2+. A decrease in SERCA2a activity and Ca2+ uptake have been shown to be responsible for abnormal Ca2+ homeostasis in both experimental and human failing hearts [33]. Associated with a defective Ca2+ uptake, there is a decrease in the relative ratio of SERCA2a/PLN in these failing hearts. With a decrease in SERCA2a expression and an increase in PLN expression, the SERCA2a/PLN ratio is significantly decreased, leading to a slower relaxation.
Using transgenic and gene transfer approaches in isolated rat cardiac myocytes and failing human myocytes, increasing levels of PLN relative to SERCA2a significantly altered intracellular Ca2+ handling by prolonging the relaxation phase of the Ca2+ transient, decreasing Ca2+ release, and increasing resting Ca2+ [7, 8, 34]. Furthermore, in neonatal rat myocytes in vitro, overexpression of SERCA2a largely “rescued” the phenotype created by increasing the PLN to SERCA2a ratio [35]. More importantly, in human cardiomyocytes isolated from the left ventricle of patients with end-stage heart failure, gene transfer of SERCA2a resulted in an increase in both protein expression and pump activity and induced a faster contraction velocity and enhanced relaxation velocity, thereby restoring these parameters to levels observed in nonfailing hearts [36]. In an animal model of pressure-overload hypertrophy in transition to failure, where SERCA2a protein levels and activity are decreased and severe contractile dysfunction is present, overexpression of SERCA2a by gene transfer in vivo restored both systolic and diastolic function to normal levels [33]. These studies provide strong evidence that overexpression of SERCA2a to rescue disturbed Ca2+ cycling and myocardial function of the failing heart is indeed possible, validating the feasibility of cardiac gene transfer in heart failure patients [37].
The recent successful and safe completion of a phase 2 trial targeting the cardiac SERCA2a has the potential to open a new era for gene therapy for heart failure [38]. In contrast to SERCA2a, most studies from hypertrophied and failing hearts have shown an increase in both NCX1 mRNA and protein [39], suggesting that enhanced NCX1 function compensates for defective SR removal of Ca2+ from the cytoplasm in the failing heart, but at the cost of further depleting the SR releasable pool of Ca2+ and further increasing the probability of arrhythmogenecity. However, the exact role of NCX1 in disease and whether it participates as a compensatory or maladaptive mechanism remains controversial.
4.3.1.2 Multi-beneficial Effects of SERCA2a
In addition to its effects on cardiac contractility, SERCA2a overexpression has other ancillary cardiovascular effects that collectively improve its therapeutic effects in heart failure which are summarized in Fig. 4.2:
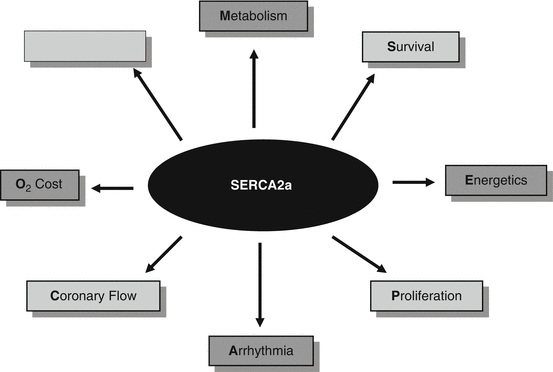
Fig. 4.2
Beneficial effects of SERCA2a overexpression. SERCA2a has various other effects on the cardiovascular system in addition to its well-established effects on myocardial contractility. SERCA2a’s effects on metabolism and energetics, survival and arrhythmia, and vascular remodeling are discussed in the text. Although individual effect of SERCA2a promotes well-established benefits, it is believed that the combination of all SERCA2a’s effects contributes to the improvement of its therapeutic effects
Metabolism and mechano-energetics: A number of studies in preclinical heart failure models demonstrated a beneficial metabolic remodeling following SERCA2a overexpression in the heart. Adeno-associated virus-mediated SERCA2a gene transfer significantly improved cardiac metabolism (as measured by the ratio of creatine phosphate to ATP) and energy utilization (as measured by restoration of the O2 consumption to EC coupling relationship to normal levels) in addition to mechanical properties [33, 40]. The normalization of myocardial energetic function by SERCA2a is an important issue because increased myocardial contractility and hemodynamics are associated with greater energy demands. This improved energy utilization by SERCA2a overexpression must have contributed to the improvement of mortality and energy reserve in heart failure [33].
Coronary outflow and myocyte hypertrophy: We have found that transcoronary gene transfer of SERCA2a increases coronary flow and reduces cardiomyocyte size in failing hearts [41]. The improved left ventricular relaxation induced by SERCA2a transfer may increase coronary blood flow via wall-dilated coronary vessels due to a more efficient relaxation because coronary blood flow occurs primarily during the diastolic phase. Increased SERCA2a activity decreases diastolic Ca2+ via increasing SR Ca2+ uptake leading to inactivation of Ca2+-regulated phosphatase, calcineurin, which plays a central role in transducing environmental signals that control gene expression and hypertrophic growth in cardiomyocytes.
Vascular reactivity: We found that human endothelial cells overexpressing SERCA2a exhibit enhanced eNOS expression and eNOS promoter activity, with increased Ser1177 eNOS phosphorylation in basal state [42]. Furthermore, we demonstrated by co-immunoprecipitation that SERCA2a and eNOS are associated in functional protein–protein complex. This indicates that both proteins are localized in a similar Ca2+ environment and also suggests that SERCA2a may directly control eNOS activity. These findings suggest that increased coronary flow occurring after intracoronary SERCA2a gene transfer in a heart failure model may be also due to increased eNOS expression and activity in coronary artery endothelial cells [41].
Proliferation: In human and animal vascular smooth muscle cells (VSMCs), proliferation is controlled by alteration of Ca2+ cycling mediated by loss of SERCA2a and RyR2 [43]. In hamsters’ VSMC, basal voltage-independent Ca2+ influx is required for activation of the nuclear factor of activated T-cell (NFAT) signaling pathway and induction of proliferation. Restoration of SERCA2a expression in proliferating cell leads to complete inhibition of basal Ca2+ influx, NFAT transcriptional activity, and proliferation of VSMC. Furthermore, SERCA2a gene transfer prevents migration of cultured VSMC. In vivo and ex vivo studies conducted in animals and humans demonstrated that SERCA2a gene transfer prevents injury-induced restenosis and vascular remodeling, inhibits inflammation in injured segments, and allows complete re-endothelization.
Ionic imbalance and arrhythmias: SERCA2a overexpression was found to decrease ventricular arrhythmias in a rodent model of ischemia–reperfusion [44] and in a large animal model of ischemia–reperfusion [45]. More recently, another group found that overexpression of SERCA2a suppresses electrical alternans, interrupting an important pathway leading to cardiac fibrillation. These findings are further discussed below. Although there were early concerns with SERCA2a overexpression because of the additional SERCA2a pumps, the SR would trigger Ca2+ release especially with leaky ryanodine receptors [46] and as such expression of SERCA2a can induce pro-arrhythmogenic effects. However, the aforementioned studies provided strong evidence that SERCA2a restoration in the setting of heart failure is rather anti-arrhythmogenic and beneficial (Chap. 48).
4.3.1.3 RyR2 and Ca2+ Regulation
It was demonstrated that RyR2 phosphorylation is highly increased in heart failure and this “hyperphosphorylation” increases the RyR2 open probability to cause a persistent leak of Ca2+ from the SR, further impairing the SR Ca2+ load. Usually, the RyR2 is closed during diastole to allow for SERCA2a Ca2+ uptake. Ca2+ release through RyR2 is regulated in part by the interaction of RyR2 with calstabin2 (also known as FKBP12.6). Protein kinase A (PKA)-induced hyperphosphorylation of RyR2 in failing hearts causes calstabin2 to dissociate from RyR2 channel complex and destabilizes the closed state of the channel, resulting in increased spontaneous diastolic RyR2 activity, elevated diastolic SR Ca2+ leak, reduced SR Ca2+ load, and decreased Ca2+ transients [1, 47]. Other suggested mechanisms for increased RyR2 open probability include oxidative modification of the RyR2 and increased RyR2 phosphorylation by CaMKII [48]. Furthermore, the gating of RyR2 is also controlled by a score of other Ca2+ regulatory proteins including triadin/junction/calsequestrin complex, sorcin, calmodulin, and the protein phosphatases PP1 and PP2A.
Diastolic leak of SR Ca2+ has been proposed as the mechanism responsible for delayed after polarization, triggered ventricular arrhythmias, and sudden death in heart failure. Although substantial data have accumulated in support of the RyR2 hyperphosphorylation hypothesis, many reports have questioned it and, using a similar canine model of heart failure (in addition to human tissue), have found that the Ca2+ sensitivity of the RyR2 opening was unaffected [49]. Furthermore, the association of calstabin2 with RyR2 has been reported to be insensitive to the degree of PKA phosphorylation. Although impaired RyR2 function is likely to be involved in the abnormal Ca2+ handling commonly observed in heart failure, the exact alterations in RyR2 function in heart failure have yet to be defined.
4.3.1.4 IP3R and Ca2+ Regulation
Another mechanism for aberrant Ca2+ signaling in heart failure is the function of inositol 1,4,5-triphosphate-gated Ca2+ release channels (IP3R) located in the SR and nuclear envelope. Although the physiological role of IP3R in cardiomyocytes has been a matter of debate, recent evidence supports a prominent role in EC coupling and SR Ca2+ release; their location near the RyR2 within the dyadic functional microdomains suggests that they can trigger CICR and enhance EC coupling [50–52]. The expression of IP3R, which is typically manyfold less abundant than RyR2 in normal cardiomyocytes [52, 53], increases significantly in both human and animal models of hypertrophy and heart failure, particularly their expression in the dyadic junction, suggesting that these channels may be associated with pathological signaling [54]. Studies in neonatal rat ventricular myocytes suggest that activation of IP3R may be linked to α1-adrenergic receptor-induced Ca2+ spark rate and global Ca2+ oscillations and catecholamine-induced cardiomyocyte hypertrophic growth. Heart-specific genetic manipulations of IP3R signaling demonstrated that IP3R expression enhances cardiac hypertrophy remodeling in vivo in response to agonists stimulation [16]. Increased IP3-induced Ca2+ release in the perinuclear region can directly activate Ca2+-dependent transcriptions factors that permit expression of hypertrophic genes [51, 55]. However, enhanced SR Ca2+ release through IP3R has the potential to trigger arrhythmogenic events [55].
4.3.2 Electrical Remodeling in Hypertrophy and Heart Failure
Although changes in many ion channel currents have been reported in cardiac hypertrophy, reduction in the density of the transient outward K+ current (I to) is the most prominent ionic current change resulting in prolongation of the action potential duration (APD) in a process referred to as electrical remodeling [56, 57]. Prolongation of the APD is consistently observed in experimental models of cardiac hypertrophy and failure. APD prolongation and downregulation of I to are also observed in compensated hypertrophy and in end-stage human heart failure [58]. In the mammalian heart, I to is encoded by Kv4.2 or Kv4.3 or a combination of the two [59]. Associated with the reduction in I to density, downregulation of Kv4.2 and/or Kv4.3 mRNA and protein expression levels has been observed in cardiac tissue derived from diseased hearts [60, 61]. APD has been shown to strongly influence Ca2+ transient amplitude in normal and hypertrophied myocytes [61] which may help to support contraction of the compromised myocardium but may also harm the myocardium by increasing the propensity to develop arrhythmias [59] and by activating hypertrophic signaling pathways (Chaps. 46 and 48).
The relationships between reductions of I to and concomitant APD prolongation and cardiac hypertrophy and heart failure have been explored using a number of techniques. Cardiac-specific ablation of I to by overexpression of a Kv4.2 channel with a single missense mutation (W362F) did not induce cardiac hypertrophy [62]. However, cardiac-specific overexpression of dominant-negative Kv4.2 channel (truncated channel: Kv4.2N) resulted in hypertrophy and cardiomyopathy along with a prolonged APD. APD prolongation is associated with an enhanced propensity to develop cardiac arrhythmias which may contribute to the high incidence of sudden death observed in patients with heart failure [59, 63].
The mechanism by which APD prolongation contributes to arrhythmogenesis might be related to the intracellular Ca2+ overload it generates. APD prolongation can elevate [Ca2+]i and several groups have demonstrated that modulation of APD is an important determinant of Ca2+ influx through LTCC. We have shown that cardiac gene transfer of Kv4.3-based I to can increase I to density, shorten APD, decrease Ca2+ influx, and attenuate cardiac hypertrophy in vitro [64] and in vivo [65]. Interestingly, restoration of the otherwise downregulated expression of the potassium channel-interacting protein 2 (KChIP2, a KV4 subunit) in hypertrophied hearts evoked similar effects [66]. Therefore, a reduction in I to density and APD prolongation represent early electrical remodeling events in the diseased myocardium, pointing toward a potential role in disease initiation and progression.
4.4 Calcium Signaling in Cardiac Arrhythmia
4.4.1 Pathogenesis and the Role of Ca2+ Overload During Cardiac Arrhythmia
Ventricular arrhythmias are a major cause of death in patients with heart failure. The role of abnormal cytoplasmic Ca2+ regulation is thought to be a critical and perhaps a common mechanism underlying cardiac dysfunction and the genesis of ventricular arrhythmias [67]. In fact the well-described phenomenon of Ca2+-mediated arrhythmias was originally observed as a result of digitalis intoxication. By raising intracellular Na+, cardiac glycosides reduce Ca2+ efflux by NCX and favor net Ca2+ uptake by the SR. At high doses, these agents can produce Ca2+ overload of the SR and result in the spontaneous release of Ca2+ by RyR2, thereby generating a net depolarizing transient inward current mediated by NCX.
Spontaneous increases in membrane potential caused by the activation of the transient inward current manifest as delayed afterdepolarizations (DADs) that can be of sufficient magnitude to produce a full-blown premature action potential. A variety of complimentary changes occurring as a result of left ventricular dysfunction in heart failure can reduce the threshold for the induction of DAD-mediated triggered beats, among which is the upregulation of NCX and the increased open probability of PKA-hyperphosphorylated RyR2 channels.
Catecholaminergic polymorphic ventricular tachycardia (CPVT) and arrhythmogenic right ventricular dysplasia (ARVD), both of which are characterized by mutations in RyR2 genes, are two forms of familial cardiomyopathy-associated arrhythmias. Wehrens et al. demonstrated that CPVT mutant RyR2 channels have a decreased binding affinity for calstabin2. Although these mutant channels are indistinguishable from wild-type channels at rest, they exhibit exaggerated calstabin2 dissociation from RyR2 in response to PKA-mediated phosphorylation, causing more SR Ca2+ release and diastolic Ca2+ leak [48].
In addition to DADs, early afterdepolarizations (EADs) are another source of triggered activity in cardiomyocytes, which is secondary to abnormal Ca2+ cycling and which results from the reactivation of LTCC during conditions of prolonged action potential repolarization. Furthermore, abnormal Ca2+ cycling by the SR has also been implicated in the pathogenesis of reentrant arrhythmias by promoting action potential alternans and/or spiral wave breakups leading to the degeneration of ventricular tachycardia and ventricular fibrillation [68, 69]. As such, changes in Ca2+ handling in general, and Ca2+ overload in particular, are thought to form the triggers for lethal arrhythmias at the cellular level, which under appropriate conditions might produce sustained arrhythmias at the organ-system level.
4.4.2 Targeting Myocardial Ca2+ Cycling to Prevent Arrhythmia
To highlight the importance of SR Ca2+ in the regulation of ventricular arrhythmias, we have addressed the issue of restoring Ca2+ load of the SR through SERCA2a overexpression using genetic strategies [44]. We and others have demonstrated that targeted delivery of SERCA2a to the heart can, as discussed above, significantly improve contractile function in vitro and in vivo, normalize metabolism and intracellular signaling pathways, and improve survival and mechano-energetic properties at no cost of O2 [33, 40, 70]. Whether these potential benefits of SERCA2a can be further extended to improve cardiac arrhythmias was addressed using a well-documented Ca2+ overload rat model of ischemia/reperfusion (IR). Cardiac IR induces ventricular arrhythmias, including ventricular premature beats, ventricular tachycardia, and ventricular fibrillation in both experimental animal models and in humans [5]. Using continuous telemetry, we found that SERCA2a overexpression significantly decreased ventricular arrhythmias during IR and 24 h later [44]. In addition, SERCA2a overexpression significantly reduced infarct size and improved wall thickening in the anterior wall which may have contributed to the decrease in ventricular arrhythmias because the size of the infarct is associated with the incidence and frequency of arrhythmias. A decrease in diastolic Ca2+ and better handling of intracellular ions during the rush of reperfusion are both associated with improved survival of the cardiomyocyte. The reduced incidence and severity of threatening arrhythmias of IR in the SERCA2a-overexpressing animals also corresponded to a preservation of muscle function, as shown by improved wall thickening and hemodynamic measurements. These findings were recently extended by other studies which demonstrated that SERCA2a gene transfer in animal models of heart failure significantly reduced arrhythmogenic cardiac alternans [71], reduced SR Ca2+ leak – through reduction of RyR2 phosphorylation – and attenuated ventricular arrhythmias in vivo [37].
Although targeting SR Ca2+ cycling proteins can abrogate Ca2+-mediated adverse electrophysiological remodeling associated with IR injury, other candidates have also been shown to exert beneficial anti-arrhythmic effects. For instance, protein kinase C epsilon (PKCε) activation confers cardioprotection from IR injury in various cell cultures, isolated perfused heart models, and transgenic mice. Furthermore, in vivo activation of PKCε was shown to protect the ischemic myocardium from reperfusion arrhythmias, while its inhibition exacerbates their incidence [72]. CaMKII has been functionally implicated in the mediation of (1) frequency-dependent acceleration of relaxation, (2) RyR2-mediated SR Ca2+ leak, and (3) Ca2+ current facilitation, which all lead to perturbations in intracellular and SR Ca2+ balances and triggered arrhythmias. In addition, CaMKII expression and activity are known to increase in patients and in many animal models of structural heart disease. Hence, its inhibition has been shown in several studies to reduce hypertrophy, cardiac dysfunction, and importantly ventricular arrhythmias [73].
4.5 Calcium Signaling in Vascular Biology
4.5.1 Role of Ca2+ Cycling in the Modulation of Arterial Vascular Smooth Muscle Cell Phenotype
Vascular proliferative disorders such as atherosclerosis and hypertension are the most common causes of severe cardiovascular diseases, the current leading cause of death in the United States. The proliferative response of VSMC is essential in injury recovery after coronary angioplasty and stent implantation. Although VSMCs are normally located in the arterial media and maintained in a contractile/quiescent state, injury or mechanical stress of arteries causes migration of VSMCs into the intima layer of the arterial wall, where the VSMCs switch their phenotype and start to proliferate and synthesize extracellular matrix proteins, resulting in expansion of the arterial intima [74]. Changes in phenotype have been well characterized at the level of contractile proteins and more recently at the level of Ca2+-handling proteins. Indeed, the LTCC are replaced by the TTCC [75], store-operated channels and TRP1 channels are upregulated [76], and a decrease in expression of RyR2 and SERCA2a in proliferating VSMC in culture has been shown [77].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


