Fig. 12.1
Schematic representation of SR Ca2+-cycling proteins and their global interactions in cardiomyocytes. (a) Calcium enters through the dihydropyridine receptor (DHPR) and activates the ryanodine receptor (RyR) to release calcium from the SR. Intracellular calcium binds to the myofibrils and triggers muscle contraction. Upon relaxation, there is reuptake of calcium by the SR Ca2+-ATPase pump (SERCA2a) or removal of the cytosolic Ca2+ by the sodium/calcium exchanger (NCX). Phospholamban (PLN), in its dephosphorylated state, binds to and inhibits SERCA2a activity. HRC interacts with SERCA2a directly and triadin (TRDN) and synchronizes SR Ca2+ uptake and release. Calsequestrin (CSQ) interacts with both TRDN and junctin (JCTN) and regulates Ca2+ release by RYR. (b) Schematic interactions of the SR Ca2+-transport ensemble, including Inhibitor-1 (I-1), type 1 protein phophatase (PP1), HS-1 associated protein X-1 (HAX1), heat shock protein 20 (Hsp20), and histidine-rich calcium binding protein (HRC). I-1, Hsp20 and PP1 are involved in the regulation of PLN activity; HAX1 interacts with PLN and possibly links SR Ca2+ cycling with cell survival
In addition, SR Ca2+ cycling is regulated by the histidine-rich calcium binding protein, HRC, which interacts with the ryanodine receptor Ca2+ release complex as well as SERCA2a [8]. Importantly, cardiac overexpression of HRC in mice inhibits SR Ca2+ uptake and cardiomyocyte relaxation [9], while HRC ablation has opposite effects [10]. Thus, HRC is a key regulator of SR Ca2+-uptake, storage and release.
Collectively, studies on the SR calcium transport regulatory proteins at the intact animal, organ, cellular, and molecular levels (Fig. 12.2) have demonstrated that these proteins are important not only in the physiological cardiac function but also in pathological conditions, and may represent promising therapeutic targets for heart disease.
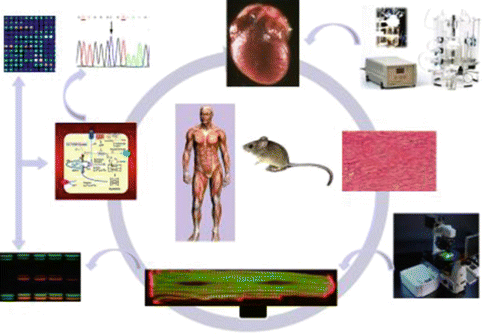
Fig. 12.2
Studies from the clinical side are extended to the bench side. Mouse models are studied at the intact animal, organ, cellular and sub-cellular levels, including genetic and molecular pathways using a wide range of approaches: echocardiography, catheterization, Langendorff perfusion, confocal microscopy, DNA sequencing, whole genome microarrays and a variety of protein assays
12.2 Role of SR Calcium Cycling Proteins and Cardiac Function Sarcoplasmic Reticulum Ca2+-ATPase (SERCA)
The 110 kD transmembrane protein, is the major regulator of Ca2+ homeostasis and contractility in cardiac and skeletal muscle. SERCA belongs to a family of highly conserved proteins and SERCA2a is primarily expressed in the heart [11]. As mentioned above, the excitation-contraction coupling in the heart is dependent on the Ca2+ reuptake function of the SR, which is mainly regulated by the SERCA2a pump. Decreases in SERCA2a gene expression levels and activity have been correlated with defects in SR Ca2+-uptake function in animal and human heart failure [12]. The role of altered SERCA2a levels in the heart has been extensively studied utilizing genetically altered models [13]. Transgenic mice over-expressing SERCA2a demonstrated an enhanced contractile phenotype [14]. Accordingly, disruption of both copies of the SERCA2a gene resulted in early embryonic lethality [15, 16], whereas heterozygous (haploinsufficient) mice with a single functional allele survived and exhibited impaired cardiac contractility and relaxation without overt heart disease, indicating that heterozygous hearts were able to meet the functional requirements under normal conditions [15, 16]. However, increased hemodynamic stress in these mice resulted in an accelerated pathway to HF, demonstrating that HF occurred more rapidly with reduced SERCA2a levels in conjunction with pressure overload [17]. Generation of the in vivo model systems provided the investigators with the possibilities to explore how changes in SERCA pump levels affected the contraction-relaxation cycle of the heart. Furthermore, these studies suggested that there was a correlation between SERCA2a levels and modulation of cardiac contractility and further supported the notion that SERCA2a function was one of the fundamental determinants of cardiac contractility and an important candidate for gene therapy approaches in failing hearts.
Importantly, a major characteristic of failing human myocardium is abnormal calcium cycling as a consequence of reduced SERCA2a expression and increased inhibition of the pump by dephosphorylated PLN [18, 13]. Although pharmacologic therapies have provided gains in reducing the mortality rates in HF, the rising incidence of the disease indicates that new and novel treatment strategies are urgently needed. Early studies had shown that increasing the activity of SERCA2a via gene transfer resulted in enhanced contractility in isolated failing human cardiac myocytes, and led to improvement in cardiac function in animal models of HF [19–21]. With the advancement of gene vectors, SERCA2a emerged as an attractive clinical target for gene delivery purposes, which is now undergoing clinical testing in HF patients. Using adeno-associated virus constructs, SERCA2a upregulation has been found to improve myocardial function in small and large animal models with HF [21, 22] and these studies paved the way for clinical trials in patients with HF [23–25]. The first-in-human gene therapy trial, Calcium Upregulation by Percutaneous administration of gene therapy In cardiac Disease (CUPID), used an adeno-associated virus carrying SERCA2a in 39 patients with New York Heart Association class III/IV HF. Treatment with the SERCA2a adenovirus resulted in improvement or stabilization in the New York Heart Association class, walking stress test, N-terminal prohormone brain natriuretic peptide levels, peak maximum oxygen consumption and left ventricular end-systolic volume, as well as decreased frequency of cardiovascular events and duration of hospitalizations [24]. More importantly, it was shown that expression of SERCA2a was still observed after 3 years on initial delivery. The long term SERCA2a transgene expression continued to exert beneficial effects in these patients with advanced HF [25]. Thus, adeno-associated virus -mediated delivery of SERCA2a appears safe and effective for treating HF in phase 1 and 2 clinical trials [23, 24]. As a result, this approach has now moved on to the phase 3 trial with a large cohort of patients [25].
Interestingly, additional targets are emerging, which may be considered as candidates for genetic manipulation in diseased hearts. Recently, it was demonstrated that SERCA2a activity could be modulated by post-translational modifications [26]. The small ubiquitin-related modifier (SUMO1) was shown to regulate SERCA2a and act as a SERCA2a-enhancing factor. This regulation seems to be essential for preserving SERCA2a ATPase activity and stability in mouse and human cell modifications [26]. Furthermore, over-expression of SUMO1 in a rodent model of heart failure had favorable effects on myocardial function. Thus, SUMOylation is a critical post-translational modification, which regulates SERCA2a function, and may provide another platform for the design of novel therapeutic strategies for HF.
In summary, increased expression of SERCA2a leads to increases in calcium handling by the SR, which appears to be a benefit that accompanies the improved contractile phenotype of the heart under pathological conditions.
12.2.1 Phospholamban Regulation of SERCA and Cardiac Function
It is well established that SERCA2a activity is directly modulated by a 52-amino acid endogenous inhibitor, PLN. The inhibitory effect of PLN on SERCA2a activity was first revealed using genetically-altered animal models. Cardiac-specific overexpression of PLN inhibited SR Ca2+ uptake and reduced systolic Ca2+ levels, contractile parameters, and basal systolic function in mice [27]. In contrast, PLN knockout (KO) mice exhibited enhanced Ca2+ cycling and myocardial contractility with no gross developmental abnormalities. The elevated contractile parameters were associated with increased affinity of SERCA2a for Ca2+ [28]. Furthermore, it was found that the hyperdynamic cardiac function in PLN-KO mice persisted through the aging process. The persistence of hyperdynamic cardiac function over the long term did not induce any pathological or adverse functional consequences, suggesting that PLN may constitute an important target for treatment in heart disease [29].
In addition, comparative analyses with wild type, heterozygous and homozygous PLN-KO mice revealed that the relative PLN levels correlated well with the affinity of SERCA2a for Ca2+ and with the rates of relaxation and contraction in cardiomyocytes, intact hearts and in vivo [27, 28, 30, 31]. These findings suggested that the PLN level may impact SR function and cardiac contractility. Indeed, gene transfer approaches to increase the levels of PLN relative to SERCA2a in isolated cardiomyocytes have been reported to significantly alter intracellular calcium handling by prolonging the relaxation phase of the calcium transient, decreasing calcium release, and increasing end-diastolic calcium concentration [32]. Accordingly, antisense strategies to inhibit PLN indicate improved sarcoplasmic reticulum function, calcium mobilization, as well as significantly improved cell shortening in cardiomyocytes isolated from failing human hearts [32, 33]. Furthermore, overexpression of a dominant negative mutant of PLN has been reported to enhance SERCA2a activity [34]. Taken together, these studies suggest that PLN may have a key role in the maintenance of SERCA2a activity and that the PLN/SERCA2a ratio is a critical determinant of basal contractility in cardiomyocytes [35].
The prominent role of PLN in regulation of Ca2+-cycling and excitation-contraction coupling was further supported by the identification of naturally occurring genetic variations in the human PLN gene, which appear to associate with heart failure. There have been four identified naturally occurring mutations in the coding region of the human PLN gene [36–39]. The human V49G point mutation was associated with super-inhibition of SERCA2a and heart failure induction in mouse models [37], while the human R9C-PLN mutation resulted in early death from dilated cardiomyopathy (DCM) in human carriers [36]. The underlying mechanisms included decreases in PLN phosphorylation and chronic inhibition of SERCA2a [36]. The T116G point mutation resulted in a stop codon (L39stop) and homozygosity was associated with DCM at a young age, while the heterozygous individuals exhibited hypertrophy without diminished contractile performance [38]. Another human mutation deleted the arginine 14 amino acid in PLN and heterozygous carriers developed heart failure by mid-age [39]. The R14del-PLN acted as a super-inhibitor of SERCA and cardiac pathology along with early death was observed in mouse models expressing this mutation in the heart [39]. Thus, there are human PLN mutations that are associated with predisposition to dilated cardiomyopathy.
12.2.2 The Anti-apoptotic Protein HAX-1 as a Regulator of Cardiac Function
The HS-1 associated protein X-1 (HAX1) was identified as a new PLN binding protein, using a human cDNA library and the yeast-two-hybrid screening approach [40]. Extensive protein studies led to the identification of the minimal binding domains of HAX1 (residues 203–245) and PLN (residues 16–22), indicating a direct physical interaction (Fig. 12.1b). Phosphorylation of PLN or elevation of the concentration of Ca2+ leads to dissociation of HAX1 from PLN, similar to findings on the PLN/SERCA2a interaction, thus indicating a physiological/pathophysiological significance of this association in cardiac muscle. Although HAX1 localizes to mitochondria, in the presence of PLN it becomes redistributed and co-localizes with PLN at the endoplasmic reticulum [40]. Analysis of the anti-apoptotic function of HAX1 revealed that the presence of PLN enhanced the HAX1 protective effects from hypoxia/reoxygenation induced cell death [40]. These findings suggest a potential link between the SR Ca2+ handling and cell survival, mediated by the PLN/HAX1 interaction.
12.2.3 The Role of Inhibitor-1 in Cardiac Function
The type-1 phosphatase (PP1) activity is significantly elevated in human and experimental HF. These increases have been suggested to be a contributing factor in the depressed cardiac function and remodeling. Indeed, cardiac overexpression of the catalytic subunit or PP1c in mouse models at similar levels as those observed in human failing hearts resulted in depressed function, remodeling, HF and early death [41]. Thus, it was suggested that inhibition of this enzyme by its endogenous inhibitor 2 and inhibitor 1 may hold promise for targeting the increased PP1 activity in HF. Gene delivery of inhibitor 2 in a rat model prevented HF progression and prolonged survival [42]. In addition, increased activity of inhibitor 1 (I- 1) exhibited therapeutic promise in HF. Inhibitor-1 is activated by protein kinase A (PKA) phosphorylation and this results in potent inhibition of PP1 and increased PLN phosphorylation, leading to enhanced Ca2+-cycling (Fig. 12.3). The role of I-1 in the heart was elucidated by genetically altered mouse models. The I-1 knock-out mouse model (KO) presented with increased PP1 activity, had a depressed cardiac function under basal conditions and attenuated β- adrenergic response [41]. These effects were associated with decreased levels of phosphorylation on Serine 16 and Threonine 17 of PLN [41]. Accordingly, overexpression of a truncated (AA: 1–65) and constitutively active (T35D) form of I-1 (I-1c) demonstrated that I-1c could inhibit PP1 activity, increase Ser16 and Thr17 phosphorylation of PLN and attenuate the hypertrophic response, delaying the onset of HF [43]. In addition, cardiac-specific and inducible expression of I-1c in the adult heart revealed that I-1c enhances basal cardiac function, which is associated with increases in PLN phosphorylation levels [44]. Furthermore, under stress conditions (transverse aortic constriction or in vivo ischemia/reperfusion), either conventional or inducible expression of I-1c was associated with increased PLN phosphorylation and increased SR Ca2+-transport. This enhanced SR calcium cycling improved the heart’s ability to accommodate the hypertrophic stimulus, delay the progression from hypertrophy to failure and impact cell survival under stress conditions. Thus, targeting I-1 may be beneficial in alleviating the detrimental effects of HF, through specific modulation of the PLN-coupled PP1 activity.
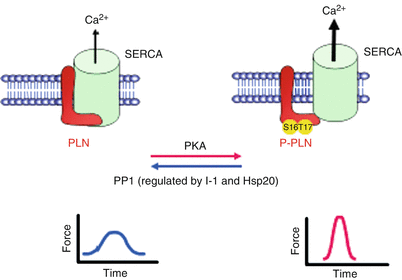
Fig. 12.3
Proposed role of Inhibitor-1 (I-1), PP1 and HSP20 in SR calcium cycling. PLN regulates the activity of the Ca2+-ATPase pump in the SR. Dephosphorylated PLN exhibits an inhibitory effect on SERCA2a. PKA- and CaMKII-dependent phosphorylation of PLN at Ser16 and Thr17, respectively, relieve the inhibition and allow for increased Ca2+ pumping into the SR. PP1 is the main phosphatase responsible for dephosphorylating PLN. Inhibitor-1 and Hsp20 suppress PP1 activity, resulting in enhanced contractility
Importantly, long-term inducible expression of constitutively active inhibitor-1, I-1c, in the heart (up to 20 months) preserved increased PLN phosphorylation and did not affect survival rates or cardiac remodeling due to chronic increases in Ca2+-cycling and function [45]. Furthermore, long-term expression of I-1c using recombinant adeno-associated virus type 9 gene transfer in rats with HF enhanced contractility and restored remodeling, associated with increased phosphorylation of PLN at Ser16 and Thr17 [45]. Thus, studies in rodent models suggest that chronic inhibition of protein phosphatase 1, through increases in I-1c, does not accelerate age-related cardiomyopathy and gene transfer of this molecule in vivo improves function and halts remodeling. These studies were subsequently extended to a large model of HF using gene transfer of I-1c. Heart failure was induced by myocardial infarction (MI) and 1 month post MI, the animals presented with severe failure, as indicated by decreases in ejection fraction, rates of contraction (+dP/dt) and relaxation (−dP/dt). Intracoronary injection of recombinant adeno-associated viral vector containing I-1c (AAV9.I-1c) prevented further deterioration of cardiac function and led to a decrease of scar size [46]. These preclinical studies indicate that cardiac overexpression of I-1c by gene transfer may improve hemodynamic function and improve cardiac remodelling. Interestingly, a naturally occurring genetic variant in I-1 (G147D), which diminishes the cardiomyocyte response to ß- adrenergic stimulation, has been also identified [47] and this indicates that human I-1 genetic variants may contribute to depressed Ca2+-cycling and functional deterioration in HF.
Overall, the studies in small and large animal models indicate that inhibition of the elevated SR PP1 activity in heart disease by active inhibitor-1, I-1c, may constitute a therapeutic strategy to rectify the disturbed Ca2+-homeostasis and function in failing hearts. Finally, the relatively small size of I-1c and its role as an inhibitor of PP1 opens the possibility of pharmacologic manipulation in addition to, or in replacement of, gene therapy.
12.2.4 Role of Hsp20 in SR Ca2+-Cycling
Heat shock proteins (HSPs) are known to enhance cell survival under various stress conditions. In the context of cardiac function, the small heat shock protein HSP20 has emerged as a key mediator of protection against apoptosis, remodeling and ischemia/reperfusion injury. Interestingly, acute increases of HSP20 levels or activity in isolated cardiomyocytes were associated with enhanced contractile parameters and Ca2+-transients [48], which were further supported by cardiac specific HSP20 overexpression in transgenic mouse hearts. HSP20 overexpression resulted in significant enhancement of cardiac function in intact animals, and in augmented Ca2+-cycling and SR Ca2+-load in isolated cardiomyocytes [5]. Accordingly, knockdown of HSP20 by antisense RNA or microRNA-320 resulted in depressed contractility [49]. The enhanced contractility by HSP20 overexpression was associated with specific increases in the phosphorylation of PLN, relieving its inhibitory effect on the apparent Ca2+-affinity of SERCA2a. Interestingly, the activity of PP1, a regulator of PLN signaling, was significantly reduced by HSP20-overexpression, suggesting that the HSP20 stimulatory effects are partially mediated through the PP1/PLN interaction. Further studies revealed an association between HSP20, PP1 and PLN (Fig. 12.1b). Specifically, there is a physical interaction between HSP20 and PP1 [50], which could be indicative of the HSP20 role in the regulation of SR Ca2+-cycling by targeting the PP1/PLN complex (Fig. 12.3). In addition, Hsp20 protects against ischemia/reperfusion-induced cardiac injury, β-agonist-mediated remodeling, and apoptosis [6, 7], introducing an additional role of HSP20 function in the heart. Thus, the enhanced contractility together with the cardioprotective role of HSP20 implies a potential dual benefit of targeting HSP20 in treating heart disease.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


