Fig. 5.1
An isolated cardiac myocyte in which sarcomeres are delineated by an antibody staining against α-actinin
Several landmarks are readily observable to delineate the sarcomere. The Z line is a complex of structural proteins that physically delimit the sarcomere. From Z line to Z line, the sarcomere is composed of regularly spaced interdigitating thick and thin myofilaments. The thick filament is primarily composed of the molecular motor protein myosin, and titin. Titin acts in part as a molecular ruler, stretching approximately half the length of the sarcomere, and provides elasticity (Granzier and Labeit 2004). Titin is anchored to the Z line via a protein complex including but not necessarily limited to telothenin and α-actinin. Myosin is a hexamer consisting of two heavy chains, two light chains, and two regulatory subunits. The hexamers are arrayed in bipolar coordination anchored to titin forming the thick filament. Myosin contains three distinct domains: the head, neck, and the tail. The head hydrolyzes ATP and directly interacts with the actin filament. The neck is a lever responsible for transducing force from the head domain. The neck domain additionally serves as a docking site for regulatory light chain proteins. The tail domain mediates the interaction with other myosin proteins in the thick filament. The myosin dense portion of the sarcomere is designated as the “A band,” which derives its name “anisotropic” pertaining to its properties in polarized light (Huxley and Niedergerke 1954).
Interdigitating with thick myofilaments are the actin-containing thin myofilaments. The thin filaments can be observed in transmission electron microscopy and are denoted as the “I band” (for Isotropic, pertaining to its properties in polarized light) of the sarcomere. Actin is attached at its barbed end to the Z line via α-actinin. The pointed ends of the actin filaments contain capping proteins and can maintain dynamic treadmilling (Littlefield et al. 2001). The cross-bridge cycling of actin and myosin is regulated by the filamentous protein tropomyosin (Tm) and the Ca2+ regulated heterotrimeric troponin (Tn) complex. The ratio of these thin-filament proteins is tightly regulated at 7:1:1 (Actin:Tm:Tn) (Tobacman 1996).
At rest, the level of intracellular Ca2+ is insufficient to saturate Tn sites. Force can be generated by increasing the likelihood of myosin cross-bridges forming with actin by increasing intracellular Ca2+, altering cross-bridge kinetics, or by increasing the Ca2+ sensitivity of the myofilament. At the myofilament level, contractile regulation is primarily regulated through a Ca2+ sensitive switch (Farah and Reinach 1995). As Ca2+ increases, regulatory proteins alter their interactions with other myofilament proteins and cause movements in the protein tropomyosin ultimately allowing for myosin heads to interact with the actin filament. Force is generated via the “power stroke.” Myosin heads bind actin readily when not sterically hindered; hydrolysis of ATP and release of inorganic phosphate results in a ratcheting motion of the myosin along the actin filament that pulls the Z-discs closer together. The binding of ATP to the catalytic domain of myosin allows for disengagement of the actin filament so the duty cycles can repeat (Warrick and Spudich 1987; Rayment et al. 1993; Ruppel and Spudich 1996).
5.5 Tropomyosin and the Steric Blocking Model
Tropomyosin is a ~40 nm long dimeric coiled-coil α-helical protein that polymerizes end to end to span the entire length of the thin filament (Xu et al. 1999). The tropomyosin dimer spans the length of seven actin molecules. Coiled-coil protein motifs are characterized by a typical seven residue helical repeat referred to as a “heptad.” Through these heptad repeats, tropomyosin has multiple highly structured interactions with the actin filament. The prototypical heptad repeat features are labeled abcdefg where “a” and “d” are hydrophobic residues and “e” and “g” are charged residues (Lehman et al. 1995; Mason and Arndt 2004; Baruaa et al. 2011). Site “d” has an especially high propensity for being an alanine residue (35 %) (Brown et al. 2001). The regular spacing of nonpolar alanine residues allows for seven discrete actin recognition sites along the length of a single Tm dimer (Sousa et al. 2013). Mutations in these key residues can lead to significant cardiac disease (Michele et al. 1999).
Tropomyosin was first speculated to have a role in regulating contractile function nearly a half a century ago; since then, advances in biophysical techniques and microscopy have enabled high-resolution discernment of how Tm sterically modulates cross-bridge formation (Brown et al. 2005). Tropomyosin can assume at least three discrete structural states defined largely by biochemical observations: blocked, closed, and open. The “blocked state” can be observed in regulated thin filaments in the absence of Ca2+. In the blocked state, myosin heads are unable to strongly bind to stereospecific sites on actin (Mckillop and Geeves 1993) (Fig. 5.2). With Ca2+-binding troponin, Tm is capable of making an azimuthal rotation of 15–25° about the actin filament into the “closed state”. This Ca2+-bound state partially exposes myosin-binding sites on actin and allows for interaction between myosin heads and the thin filament. With the addition of strong myosin binding, Tm performs an additional movement along the actin filament to approximately 35° away from the Ca2+-free blocked state to define the open state (Spudich et al. 1972; Parry and Squire 1973; Pirani et al. 2006; Poole et al. 2006). The exact nature of how Tm moves along the actin filament is still debated, whether it is a flexible rolling or a rigid sliding maneuver (Holthauzen et al. 2004; Li et al. 2010).
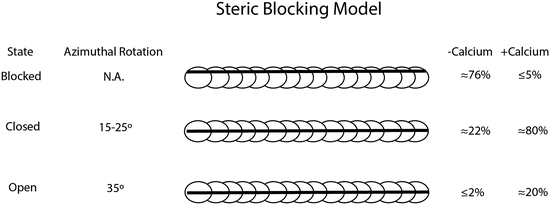
Fig. 5.2
The steric blocking model showing the relative rotational azimuthal movements of tropomyosin about the actin filament and the approximate percentage of populations occupying each biophysical state with and without calcium bound to troponin (right panel) (McKillop and Geeves 1993)
Critical to the position and movement of tropomyosin is the conformation of the heterotrimeric troponin complex. Tropomyosin is held in its blocked conformation primarily by the action of the N-terminal region of troponin T and by two regions of troponin I known as the “inhibitory region” and the “mobile domain.” As Ca2+ binds to troponin C, the equilibrium shifts between populations in the steric blocking model. During diastole, low Ca2+ levels and weak myosin cross-bridges denote a dynamic equilibrium that rests predominantly in the blocked and closed states. During systole, the influx of Ca2+ and the positive feedback of strong myosin head attachment shift the equilibrium to chiefly the open state (Fig. 5.2).
5.6 Troponin: The Ca2+ Switch
Ebashi first discovered troponin in 1963 (Ebashi et al. 2008). At the time, it was recognized as “native tropomyosin,” a tropomyosin-like protein that participated in the Ca2+ binding and relaxing action of muscle. In the over 50 years since its discovery, our understanding of troponin has grown from being another protein on the list of muscle regulators to being the master molecular switch complex that mediates the link between excitation and contraction (Ebashi 1963; Farah and Reinach 1995).
Troponin is a heterotrimeric complex of the proteins troponin I, C, and T. In striated muscle, the binding of Ca2+ to troponin regulates contraction (Farah and Reinach 1995). In the absence of troponin, muscle will contract independent of Ca2+ (Moss et al. 1986). The troponin complex is anchored to the thin filaments by multiple protein–protein interactions between TnI, TnT, Tm, and Actin. Troponin I is named for its inhibitory effects on actin/myosin ATPase; troponin C is named for its Ca2+-mediated regulation of thin-filament conformational dynamics and troponin T for its role in anchoring the heterotrimeric complex to tropomyosin. Troponin functions to convert Ca2+ signaling into biophysical movements that dictate force generation. In 2003 the core domain of the troponin complex, which houses most of the regulatory function, was resolved to 2.6 Å resolution via X-Ray crystallography. This significant advance represented the first time the heterotrimeric troponin complex was visualized to atomic level resolution (Takeda et al. 2003).
5.7 Troponin C
Troponin C (TnC) is often described as a dumbbell shaped protein with two globular domains connected by a central linking helical domain. TnC is an approximately 18 kDa protein derived from two distinct genes (Fig. 5.3). TNNC1 encodes for the TnC isoform expressed in slow skeletal (ssTnC) and cardiac muscle (cTnC), whereas TNNC2 encodes for the fast skeletal isoform (fsTnC) found in fast twitch limb muscles. TnC belongs to the EF-hand domain family of proteins, characterized by their canonical helix-loop-helix divalent metal-binding sites. Each globular domain of troponin C contains two EF hand domains, from N-terminus to C-terminus the sites are numbered I, II, III, and IV (Herzberg and James 1985) (Fig. 5.3). Sites III and IV in the C-terminal globular domain are high affinity cation sites and constitutively bound to Ca2+/Mg2+; these are generally considered structural domains. In contrast, the N-terminal sites are regulatory. Despite 70 % sequence homology to the skeletal isoform, site I in the cardiac isoform is inactive due to seven key loop residue substitutions; subsequently, all cardiac Ca2+ responsiveness is conferred via site II (Van Eerd and Takahashi 1975). Troponin C can functionally be classified as a signaling protein, as binding of Ca2+ to site II causes dramatic conformational changes not only in troponin C but also the troponin complex and tropomyosin to activate the thin filament and ultimately generate force.
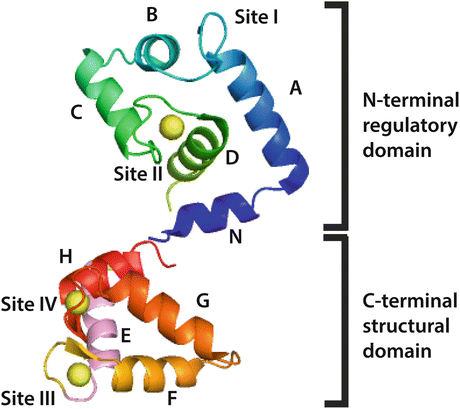
Fig. 5.3
Ribbon structure schematic of cTnC (PDB 1J1E) in the Ca2+-saturated state with EF hands and critical helix domains labeled. In this structure, the central helix that links the DE helices is not resolved. Note that Site I is inactive in cTnC
Much of the groundwork for understanding the conformational dynamics of cardiac troponin C is derived from the understanding of the fast skeletal isoform (fsTnC). X-ray crystallographic studies of the skeletal troponin from avian species suggest the elongated dumbbell shape of troponin C, where the two globular domains are connected by a central helix (Herzberg and James 1985; Satyshur et al. 1988). These crystal structures allowed a more intimate understanding of the conformational changes that occur upon Ca2+ binding to TnC (Strynadka et al. 1997). It was purported that binding of Ca2+ to the regulatory site would cause helices A and D of troponin C to move relative to helices B and C (see Figs. 5.2 and 5.3). This movement would expose hydrophobic residues of the N-terminal domain and allow for Ca2+-dependent interaction with troponin I (Herzberg and James 1985; Sundaralingam et al. 1985; Mehler et al. 1991; Gagne et al. 1995). It is important to note that in the cardiac isoform, complete exposure of the hydrophobic residues requires troponin I in addition to Ca2+ (Gagne et al. 1995).
High affinity Ca2+-binding sites III and IV (KCa ~ 1.0 ± 0.2 × 107 M−1) are sensitive to competitive binding to magnesium (KMg ~ 3.0 × 103 M−1), unlike the lower affinity Ca2+-binding site II (KCa ~ 2.5 ± 1.6 × 105 M−1). When TnC is complexed in either the dimeric complex with TnI or the native Tn complex, the Ca2+ affinities increase for both the non-regulatory and regulatory sites (KCa ~ 3.2 ± 1.2 × 108 M−1, and KCa ~ 2.5 ± 2.0 × 106 M−1, respectively) (Holroydes et al. 1980). This reflects the enhanced Ca2+ sensitivity of troponin C as thin-filament complexity increases.
Initially, it was thought that the rate of Ca2+ exchange with TnC was faster than the kinetics of contraction and relaxation in cardiac muscle. Kinetic studies of troponin C in isolation showed Ca2+ off rates between 590–995 s−1 at 4 °C and 1,000–1,263 s−1 at 15 °C using a variety of methodologies (Dong et al. 1996; Tikunova and Davis 2004). However, given the change in Ca2+ affinity at all sites when cTnC is in its native complex, it is unlikely that these rates reflect the true nature of Ca2+ dissociation in situ. Indeed, Ca2+ dissociation also slows as thin-filament complexity increases; with the addition of cTnI to cTnC dissociation of Ca2+ slows from >1,000 s−1 at 15 °C to 122 s−1 at 15 °C. With the addition of cTnT, Ca2+ dissociation slows further to 34 s−1 at 15 °C. It is worth noting that the addition of ssTnI to cTnC has an even more dramatic effect on Ca2+ dissociation lowering the off rate to 9.8 s−1 at 15 °C (Gomes et al. 2004). More recently, the introduction of a novel cysteine residue (T53C) for the covalent modification of fluorophores in the BC subdomain of the N-terminal globular domain of cTnC has allowed for detection of Ca2+ binding without perturbing biological function or reliance on a cytosolic Ca2+ reporter (e.g., BAPTA, Quin-2) (Davis et al. 2007). The use of this system has revealed similar off rates for the Tn complex at 15 °C (41.9 s−1). Additionally, it has been shown that the addition of tropomyosin may further slow Ca2+ dissociation to 36 s−1. Addition of actin to the Tn:Tm complex enhanced cooperativity (n = 0.91 vs. n = 1.65) of Ca2+ binding and increased Ca2+ dissociation to 105 s−1. Introduction of myosin S1 to the system had drastic effects on Ca2+ dissociation, slowing its off rate to 13 s−1 (Davis et al. 2007). Taken together this evidence suggests that unlike isolated cTnC in solution, Ca2+ dissociation from cTnC in the complex thin filament is compatible with rates of mechanical relaxation.
5.8 Troponin I
Troponin I (TnI) is an approximately 24 kDa protein encoded by three distinct genes. TNNI1 encodes for the slow skeletal isoform (ssTnI), TNNI2 encodes for the fast skeletal isoform (fsTnI), and TNNI3 encodes for the cardiac isoform (cTnI). Troponin I interacts with most other thin-filament proteins (TnC, TnT, Ac, and Tm). In this way, TnI acts as an adapter protein to facilitate the complex conformation dynamics of the thin filament. Here, cTnI is named for being the subunit of the troponin complex responsible for inhibition of actomyosin MgATPase. The immunological detection of cTnI has been used extensively in clinical settings for the specific determination of myocardial injury (Adams et al. 1993). Unlike cTnC, whose structure has been elucidated with high atomic resolution, the crystal structure of cTnI, free in solution, is not yet available. Despite this, cTnI has been the subject of extensive biochemical investigation and contains several noteworthy functional domains. The N-terminus of cTnI contains a 32 amino acid extension that is unique to the cardiac isoform. The C-terminal region contains several well-characterized regions important for the Ca2+-dependent inhibition of myosin cross-bridge cycling. Residues 128–147 (excluding the initial methionine) contain the minimal peptide region responsible for inhibition of actomyosin ATPase activity; this region is typically referred to as the aptly named “inhibitory region” (Farah and Reinach 1995). This peptide is derived from the cyanogen bromide digest of the full length protein. From this region, 12 residues (136–147) alone can recapitulate troponin-mediated thin-filament activation/inhibition (Talbot and Hodges 1981; Lindhout and Sykes 2003). Residues 148–163 contain a region which binds to the N-terminal region of cTnC upon Ca2+ binding and facilitates in the full opening of the hydrophobic patch. Residues 167–210 contain a region referred to as the “mobile domain,” with residues 168–188 containing a second actin-binding domain.
5.9 Troponin T
There are three distinct genes in the human genome that encode for troponin T. TNNT1 encodes for the slow skeletal isoform, TNNT2 encodes for the cardiac isoform, while TNNT3 encodes for the fast skeletal isoform. From each of these genes numerous isoforms are derived from various splice variants; in total there are at least 21 isoforms that have been recognized (UniProt 2014). There are four different isoforms expressed in the heart, with three isoforms being expressed in the fetal heart (and potentially reexpressed in the failing heart as part of a fetal reprogramming) and one isoform in the adult heart (Anderson et al. 1991). The four cardiac isoforms are numbered cTnT1 through cTnT4 in order of decreasing molecular weight. Troponin T (TnT) is an approximately 35 kDa protein that anchors the heterotrimeric troponin complex to the thin filament via tropomyosin. Cardiac troponin T (cTnT) is homologous amongst species with the exception of a hypervariable N-terminal region (residues 1–79). TnT interacts strongly with troponin I through a region termed the “I-T arm” (residues 234–284). In addition to its role in anchoring troponin, TnT also interacts with all major components of the thin filament including tropomyosin which interacts with two distinct binding regions (residues 89–127 and 215–240) (Wei and Jin 2011). Through changes in fiber type specific isoform regulation, posttranslational modifications, and splice variants, troponin T is able to participate in the modulation of striated muscle contraction and relaxation.
5.10 Phosphorylation
β-adrenergic stimulation via activation of the sympathetic nervous system is a predominate mechanism by which the heart rapidly responds to stress and changes in circulatory demand. The heart is able to adapt to increased demand through the summed combination of inotropic, chronotropic, and lusitropic effects. β-adrenergic signaling is primarily thought to activate cAMP-activated protein kinase (PKA) which has numerous targets that modulate both Ca2+ entry and myofilament responsiveness to Ca2+ (Metzger and Westfall 2004). This response is achieved through the PKA-dependent phosphorylation of L-type Ca2+ channels, ryanodine receptors phospholamban, and cTnI. PKA-mediated phosphorylation increases Ca2+ currents through the L-type Ca2+ channels and ryanodine receptors (this is achieved secondary to SERCA disinhibition) (Yoshida et al. 1992). PKA-mediated phosphorylation of phospholamban reduces its ability to attenuate sarcoplasmic reticulum Ca2+ ATPase (SERCA) activity, thereby enhancing Ca2+ loading kinetics and increasing total SR Ca2+ content (MacLennan and Kranias 2003). However, this review will focus on the PKA-mediated effects on cTnI.
PKA phosphorylates cTnI at serine residues 23 and 24. Residues 23/24 are part of the unique 32 amino acid N-terminal extension found exclusively in the cardiac isoform of Troponin I (Zhang et al. 1995). Phosphorylation at these residues reduces Ca2+ sensitivity of the myofilaments; that is, it causes a rightward shift in the force-pCa relationship (Yasuda et al. 2007). Reduction of myofilament Ca2+ sensitivity and enhanced Ca2+ dissociation from cTnC result in accelerated relaxation of the myofilament (positive lusitropy) (Zhang et al. 1995; Yasuda et al. 2007).
The use of gene transfer and transgenic mice harboring mutations in key residues of cTnI has been essential in elucidating the mechanistic nature of these physiological phenomena. Chimeric TnI molecule structure–function studies were critical in resolving the key functional domains of TnI (Westfall et al. 2000, 2001). Here it was shown the C-terminal domain of TnI contains the key effector residues for affecting Ca2+ and pH sensitivity inherent in the TnI isoforms. Studies in live myocytes transduced with adenovirus expressing either the slow skeletal isoform of troponin I (ssTnI), which lacks the unique N-terminal extension of the cardiac isoform, or a chimera isoform with the N-terminus of ssTnI and the C-terminus of cTnI demonstrated definitively that the N-terminal extension of cTnI is responsible for mediating the Ca2+ desensitizing effects of PKA following β-adrenergic stimulation (Westfall et al. 2001). In 2002, generation of a transgenic mouse by Pi et al. expressing cTnI where residues S23/S24 were mutated to non-phosphorylatable alanines corroborated previous evidence that these tandem series in the N-terminal extension in particular were responsible for the β-adrenergic response (Pi 2002; Pi et al. 2003). Conversely, mice have also been generated which mimic constitutive phosphorylation at serines 23/24 by mutation to aspartate. These mice show enhanced relaxation even at high heart rates that could be normalized by use of β-adrenergic agonists (Takimoto et al. 2004). Perhaps the most convincing evidence that the lusitropic effects of PKA are primarily manifested through cTnI and not other targets comes from a series of experiments in transgenic mice harboring mutations in troponin I where serines 23/24 were replaced by phosphomimetic aspartate residues. In cardiac myocytes explanted from these mice, relaxation times were significantly faster at baseline compared to control mice (Yasuda et al. 2007). Additional treatment with β-adrenergic agonists only caused nominal increases in the relaxation speed, whereas the Ca2+ transient decay rate was comparable between both transgenic and control mice treated with β-adrenergic agonists (Yasuda et al. 2007). Taken together, this evidence suggests that the PKA-modulated lusitropy involves a key role of the myofilaments and in particular cTnI.
In addition to its lusitropic effects, PKA-mediated phosphorylation also increases the inotropic state of cardiac muscle. Part of this effect is mediated through increased Ca2+ loading of the sarcoplasmic reticulum subsequent to phospholamban phosphorylation thereby increasing SERCA activity. The end result from this is larger Ca2+ amplitudes thereby increasing myofilament activation. However, independent of this effect, phosphorylation of troponin I appears to enhance function. PKA treatment of skinned rat cardiac myocytes has shown an increase in maximal force and peak absolute power output during maximal Ca2+ activations; this increased power output may be explained by the increased cross-bridge cycling rate that occurs with β-adrenergic stimulation (Kentish et al. 2001). Additional support for the notion of PKA phosphorylation mediating positive inotropic effects via the myofilament comes from the observation that transgenic mice with the S23/24D mutation have increased systolic function at baseline and force-frequency response that is independent of the SR reconciled inotropic effects (Yasuda et al. 2007).
In addition to being a target of PKA, the thin filament also serves as substrate for protein kinase C (PKC). There are 11 identified isoforms of PKC that can be classified as conventional (α, β, βII, γ), novel (δ, ε, η, θ), and atypical (ζ, λ, ι) (Steinberg 2012). In cardiac myocytes, PKC is typically activated downstream of G-protein coupled receptors (GPCRs) by the agonists: endothelin I, angiontensin II, norepinephrine, and epinephrine. Binding of agonists to GPCRs causes the activation of phospholipase C (PLC) which causes the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) to membrane-bound diacyl glycerol (DAG) and inositol triphosphate (IP3). IP3 can cause the release of internal stores of Ca2+, which along with DAG activate conventional PKCs (Steinberg 2012). Both negative and positive isotropic and lusitropic effects have been reported in cardiac mycoytes, so it is likely that there are isoform specific as well as both myofilament and non-myofilament responses to PKC activation (Westfall and Borton 2003). For this reason, we will focus on the role of PKC in phoshorylating troponin I.
Similar to studies with PKA, to discern the effects of PKC on one substrate versus another, the most effective methods have been using in vitro reconstituted skinned myocytes, acute gene transfer, and transgenic mouse models with constitutive phosphorylation mimetics. The use of PKC agonists has historically been confounded by off target effects when trying to assess the physiological consequences of thin-filament phosphorylation. Troponin I can be phosphorylated by PKC at sites S23/S24, S44/S46, and T144 (Noland et al. 1989). Phosphorylation of S44/46 causes a reduction in the maximal Ca2+-activated force, MgATPase activity, maximal sliding velocity, and the cross-bridge cycling rate (McClellan et al. 1996). PKC phosphorylation of T144 results in a rightward shift in the pCa-filament sliding speed relationship. PKC-α preferentially phosphorylates sites S44/46. Phosphorylation of these serines is common among all PKC isoforms that target TnI except PKC-β which seems to preferentially phosphorylate S23/24 and T144 (Wang et al. 2006). Despite the differing accounts of the physiological role of PKC, there seems to be consensus in the literature that phosphorylation of troponin I at sites S44/46 reduces maximal Ca2+-activated tension and T144 reduces Ca2+ sensitivity of sliding velocity which in concert may offer therapeutic targets in failing myocardium.
Troponin T has numerous putative PKC phosphorylation sites including T204, S208, T213, S285, and T294 (Noland 1996; Sumandea et al. 2009). It is apparent that cTnT is phosphorylated in vitro by various PKC isoforms; however, the studies investigating isoform specificity have returned conflicting results with some studies suggesting PKC-α and PKC-δ preferentially phosphorylated cTnI over cTnT whereas PKC-ζ was cTnT specific, while others reported the exact opposite relationship (Noland 1996; Pi 2002). Despite this, studies using phosphomimetics or reconstituted in vitro phosphorylated troponin T tend to have consensus that phosphorylation of troponin T results in reduced binding affinity of troponin T for tropomyosin resulting in a leftward shift in the pCa-actomyosin MgATPase relationship and decreases maximal tension (Noland and Kuo 1991, 1992).
In addition to posttranslational modification by the better-characterized PKA and PKC effects, troponin I also serves as substrate for several other kinases in cardiac myocytes. PKD has been shown to phosphorylate TnI at the same sites as PKA at Ser22/23 and causes a rightward shift in the tension-pCa relationship. The exact contribution by PKD in vivo is unknown, but it is believed to complement the modification of troponin I’s phosphorylation state (Haworth et al. 2004; Cuello et al. 2007). Rho-associated protein kinase (ROCK) is also capable of posttranslational modification of the thin filament. ROCK treatment of myocytes causes an inhibition of tension and ATPase activity. Sites S23/24 and Thr144 of troponin I and Ser278/287 of troponin T are all substrate for ROCK (Vahebi et al. 2005). Although their exact physiological significance is still debated, ROCK kinases are known downstream agents of stress, hypertrophy, and heart failure signaling cascades. AngII has been used to cause hypertrophy in myocytes associated with activation of the RhoA pathway (Aoki et al. 1998). p21-activated kinase (PAK) is also capable of phosphorylating TnI and TnT as well as other sarcomeric proteins. PAK phosphorylation causes an increase in peak isometric force and Ca2+ sensitivity; however, its physiological significance remains elusive (Buscemi 2002).
5.11 Ischemic Insult and the Thin Filament
Ischemic heart disease is the leading cause of death worldwide accounting for as much as 12 % of all deaths (Finegold et al. 2013). Acute ischemia bouts are typically initiated by the disengagement of an atherosclerotic plaque into the coronary vasculature that begins a clotting cascade resulting in the partial or complete occlusion of the coronary vasculature (Davies and Thomas 1985). During bouts of ischemia, left ventricular pressures will rapidly drop due in part to the accompanying biochemical changes of the myocyte (Lee and Allen 1991). It is well established that ischemia causes biochemical changes in myocytes that can result in damage to organelles, namely the nuclei and mitochondria. As oxygen becomes depleted in the cardiac myocytes, metabolic changes from normal oxidative respiration to strictly anaerobic glycolysis cause increased production of protons lending to acidification. There is increasing evidence that ischemic insult causes modifications to the cardiac cellular machinery secondary to acidification.
Which proteins are modified, degraded, or lost may be in part dependent on the extent in which the tissue is ischemic (Rajabi et al. 2007). In a canine model, occlusion of the left descending coronary artery for 15 min resulted in disruption of the microtubule network that was reversible after an hour of reperfusion and could be halted with Ca2+ chelators suggesting a Ca2+-dependent role in the disarrangement (Sato et al. 1993). α-actinin has also been shown to be highly susceptible to ischemia-induced degradation; α-actinin and troponin I were shown to be two of the more susceptible proteins to proteolysis subsequent to ischemic injury (Van Eyk et al. 1998). The alterations to α-actinin and troponin I may in part explain the observation of a time-dependent reduction in maximum force from skinned fiber bundles. The extent and functional significance of these degradations is debated among groups; however, there is consensus that these changes lead to contractile dysfunction.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


