Figure 28–1.
Four ECG traces of a resuscitated Brugada syndrome patient showing most severe ST-T abnormalities in leads positioned over the second and third intercostal space (right two panels) where a coved-type ECG is present (arrows). Intermediate ST-T abnormalities (saddleback-type) are recorded in the fourth intercostal space (leads V2–V3). Calibrations are given (Courtesy of Dr. Wataru Shimizu)
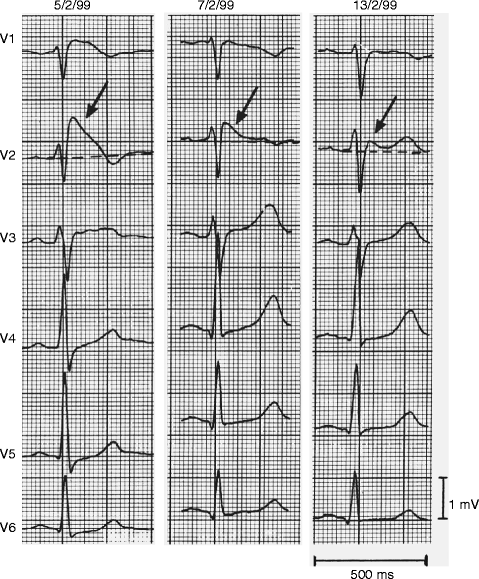
Figure 28–2.
Precordial leads ECG of a resuscitated patient with three types of ST segment elevation described in Brugada syndrome. Within a few days the ECG changed from type I (left panel) to type II (middle) and type III (right panel). For explanation see text. The arrows indicate the J waves. Calibrations are given (Modified from Wilde et al. [23])
Of these criteria, only a history of documented VT/VF has clear prognostic implications. Spontaneous occurrence of a type I ECG also has prognostic implications, representing a condition with increased risk for malignant arrhythmias and, if present in conjunction with syncope (otherwise unexplained), conferring an indication for implantation of an implantable cardioverter/defibrillator (ICD) [25, 26]. In contrast, a positive family history of SCD does not confer a higher risk to the family members, remaining therefore, only a useful criterion for the diagnosis, but not for the prognosis of Brugada syndrome patients [10, 27]. Risk stratification and indications for ICD implantation are extensively discussed in the Second Consensus Report paper, published in 2005 [24]. Since then, new data emerged which support the notion that EPS is a poor event predictor [10]. Moreover, there is increasing recognition that, in most patients, the prognosis of Brugada syndrome is not as unfavorable as initially thought. This is leading to more carefulness in ICD implantation strategy. This topic and other important aspects about management in Brugada syndrome are reported in Chap. 29. Brugada syndrome is inherited as autosomal dominant trait. It was linked to mutations in the SCN5A gene, encoding the α-subunit of the cardiac sodium channel protein [28]. Up to now more than 200 SCN5A mutations correlating with the Brugada syndrome phenotype have been found [29] (see also http://www.fsm.it/cardmoc/) (Fig. 28.3). All Brugada syndrome-linked mutations cause a reduction in sodium inward current (INa+) during phase 0 of the cardiac AP [30]. Interestingly, the type of SCN5A mutation may influence the severity of the phenotype [31], although genotype-phenotype correlation studies have been limited due to the low prevalence of mutation carriers and to the fact that only a few mutants have undergone functional studies. Also, the genetic background of an individual, i.e., the presence of polymorphisms (either on the same or other genes) that can act as modifiers, may explain the differences in severity of phenotype [32–34]. A mutation in the SCN5A gene is actually found in up to 22% of all Brugada syndrome cases [8]. Some investigators have reported that, in another 11–12% of the cases, mutations in calcium channel genes (CACNA1C, CACNB2B and CACNA2D1) are found [35, 36]. More rarely, genes encoding for sodium channel β-subunits or genes affecting transient outward current Ito current are involved [37–40]. Occasionally, the glycerol-3-phosphate dehydrogenase 1-like gene (GPD1L) is involved. Yet, with the exception of a large Dutch family carrying mutation 1795insD in the SCN5A gene [41] and of mutations in GPD1L, classic genetic linkage analysis in Brugada syndrome is lacking [42]. Furthermore, two groups showed that in several large SCN5A-related Brugada syndrome families, affected subjects (phenotype positive) did not carry the familial mutation (genotype negative) [43, 44]. For these reasons, interpretation of genetic data in Brugada syndrome patients requires extreme caution [8].
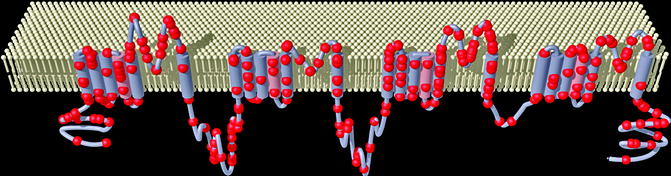
Figure 28–3.
Representation of the α-subunit of the voltage gated SCN5A sodium channel showing the locations of the mutations associated with Brugada syndrome (circle) (Courtesy of Dr. Andre Linnenbank, Department of Experimental Cardiology, Academic Medical Center, Amsterdam, The Netherlands)
General Clinical Properties
Demography
Since its recognition as a distinct subgroup of idiopathic VF in 1992, Brugada syndrome is increasingly described worldwide, although its exact prevalence remains unclear and can vary significantly between different regions of the world [45, 46]. Moreover, the Brugada syndrome ECG in the medical literature is often used as synonymous of Brugada syndrome, which is not completely correct [47, 48], since more clinical criteria need to be fulfilled for the diagnosis in addition to ECG abnormalities [23]. The Brugada syndrome is endemic in East and Southeast Asia, where it underlies the Sudden Unexplained Nocturnal Death Syndrome (SUNDS) [49]. It is also particularly prevalent in Japan, the Philippines and Thailand, being the leading cause of nontraumatic sudden death among young men [50, 51]. In China and Korea, the reported incidence is lower [52–54]. In Europe, although Brugada syndrome is less prevalent than in Asia, it is still extensively described [25, 55], with exception of the Scandinavian countries. Its prevalence is estimated at 5–50 cases per 10,000 inhabitants [56, 57]. Conversely, occurrence of Brugada-type ECG and/or Brugada syndrome in the United States and Canada seems to be uncommon, but it might be under-recognized [58–60]. In Iran, the reported prevalence of the typical Brugada syndrome ECG among subjects presenting with palpitations is greater than in some European countries, but lower than in Japan [61]. Finally, Brugada syndrome is also described in North Africa, with a prevalence of 2 per 100,000 adults, although this data may represent an underestimation [62].
Clinical Characteristics and Types of Arrhythmia
The clinical presentation is highly heterogeneous and may include palpitations, dizziness, syncope, and (aborted) sudden death. In the areas where Brugada syndrome is endemic, there seem to be also a more severe clinical course with a high incidence of VT/VF at young age [63]. Still, most subjects in whom a Brugada syndrome type I ECG is found remain asymptomatic [10, 64]. Syncope is by far the most important symptom because its occurrence has a recognized prognostic value [10], especially when associated with spontaneous segment ST elevations [26]. An estimated 80% of patients with documented VT/VF have a history of syncope [55]. Syncope may be provoked by self-terminating VT and this may explain why patients experience agonal respiration at night after which they wake up [65–69]. Interpretation of syncope episodes requires caution since several descriptions point out that syncope in subjects with a type I Brugada syndrome ECG may also present with typical vasovagal features [70, 71]. Indeed, the prevalence of a positive head-up tilt test reached 35% in two small Brugada syndrome cohorts and was higher than in control subjects [72, 73]. This suggests that the impairment of the autonomic nervous system may play a role in triggering syncopal episodes in Brugada syndrome subjects. However, a protocol on management of syncope episodes in patients suspected to have Brugada syndrome is missing and future studies on this topic are certainly needed. In any case, episodes of syncope in a young or middle aged subject should lead to screening for Brugada syndrome, especially when syncopes are recurrent and severe [74]. Arrhythmic events in Brugada syndrome can occur at all ages, from childhood to the elderly (range 0–77 years) [53, 75–77], with a peak around the fourth decade [78]. In children, clinically manifest Brugada syndrome is less prevalent than in adults [79], and a typical Brugada syndrome ECG pattern rarely appears before adolescence [80].
It has been estimated that Brugada syndrome causes 4–12% of all SCD, and up to 20% among patients without identifiable structural abnormalities [81]. However, in some families the clinical course is more benign [64]. Correct diagnosis of the disease by the index patient and prompt identification of (pre-symptomatic) affected family members is necessary in order to prevent more cases of SCD. An extensive familial examination of (aborted) sudden unexplained arrhythmic death (SADS) cases conducted at our institution unmasked inherited cardiomyopathies or channelopathies in up to 40% of the tested families [82]. According to recent reports, at least 10% of SADS families associate with Brugada syndrome [83]. Sudden death in Brugada syndrome results from fast polymorphic VT which usually originate from the right ventricular outflow tract (RVOT) [84], and degenerate into VF [1, 2, 85], whereas no significant variations in QTc intervals precede spontaneous VF episodes [1, 86]. Ventricular arrhythmias and (aborted) sudden death in Brugada syndrome- distinct from arrhythmogenic right ventricular cardiomyopathy (ARVC)- typically occur at rest when the vagal tone is augmented [87], and often at night [88,89] or after large meals [90, 91]. During the night, Brugada syndrome patients suffer (more often than controls) from sleeping disorders with episodes of apnea, which can activate the parasympathetic nervous system and function as an arrhythmogenic trigger [92].
Clinical presentation with sustained monomorphic VT, although very uncommon, has also been described [93–95], also in one infant [96]. Data obtained from stored electrograms of ICDs have demonstrated that, although premature ventricular complexes (PVCs) in patients affected with Brugada syndrome are rare [97], their prevalence increases prior to spontaneous VF [86]. These PVCs appear to have the same morphology as the first VT beat, and different VT episodes are initiated by similar PVCs in the same subject [86, 98]. They show a left bundle branch block morphology [99] and endocardial mapping localized their origin in the RVOT [100]. Further confirmation of the role of these initiating PVCs and of the RVOT derives from the clinical benefit resulting from their elimination via catheter ablation [100]. More recently, a study in nine severely symptomatic Thai Brugada syndrome patients with recurrent ICD discharges upon VF demonstrated the presence of abnormal ventricular fragmentation at the epicardial site of the anterior RVOT region [101]. After ablation in this area, the ECG normalized in the majority of the patients and no recurrence of VT/VF occurred in 8/9 patients during a follow-up of 20 ± 6 months.
This elegant publication strongly supports the hypothesis that Brugada Syndrome is caused by conduction delay in the right ventricular outflow tract.
Supraventricular Tachyarrhythmias
Episodes of atrial flutter/fibrillation (AF) are often documented in Brugada syndrome [102–106] with a prevalence up to 30% [88, 107, 108]. This is abnormally high considering the young average age of the affected patients [109]. There seems to be no difference in occurrence of AF in SCN5A mutation positive patients compared with SCN5A mutation negative patients [110]. Given that a history of atrial arrhythmias correlates with VT/VF inducibility during EPS, and that ST segment elevation correlates with the onset of AF episodes [88], Brugada syndrome patients with paroxysmal atrial arrhythmias may constitute a population at higher risk with a more advanced disease state [109], but these data are still limited [111]. Importantly, it is known that the annual incidence of inappropriate ICD shocks due to supraventricular tachyarrhythmias in Brugada syndrome patients may be as high as 14% [112–114]. Therefore, correct management strategies aiming not only to avoid ICD implantation in low risk patients but also to prevent inappropriate ICD discharges (drug-achieved and/or through accurate ICD programming) are mandatory [9, 115].
Gender Disparity
A salient property in the clinical manifestation of Brugada syndrome is the higher disease prevalence in males (70–80% of all affected subjects), particularly in regions where this syndrome is endemic, despite equal genetic transmission among both genders [50, 56, 78]. That a role in gender disparity could be played by sex hormones, in particular by testosterone, was suggested by the demonstration that castration attenuated ST elevations in two asymptomatic male Brugada syndrome patients [116] and by the revelation that men affected with Brugada syndrome have significantly higher levels of testosterone than age-matched control subjects [117]. Also, is has been shown that male subjects with Brugada-like ECG have a higher risk for prostate cancer, independently of their smoking habit, age or radiation exposure (the study population consisted of survivors of the Nagasaki atomic bomb) [118]. A possible explanation for this phenomenon, derived from clinical [119] and experimental studies [120, 121], is that sex hormones may modulate potassium currents (e.g., Ito) during the early repolarization phase of the cardiac AP.
Among affected patients, men and women differ in their clinical presentation. This is reflected in the greater amount of ST segment elevation in men (on average), while women show more pronounced conduction disorders in response to drug challenge [122]. Still, exceptions have been described [123]. Also, the prognosis seems to be worse in men in comparison with women [124], although male sex per se was not a statistically significant predictor of events in multivariate analysis in a large cohort of asymptomatic patients [10].
Genetic Aspects
In 1998 Brugada syndrome was linked to mutations in the SCN5A gene, encoding the pore-forming α-subunit of the human cardiac sodium channel protein [28]. Nowadays, 11 different genes are associated to the Brugada syndrome, with SCN5A still being the most important involved gene [8]. Yet, with the exception of a large Dutch family carrying mutation 1795insD in the SCN5A gene [41] and of mutations in the glycerol-3-phosphate dehydrogenase 1-like gene (GPD1L) [125], classic genetic linkage analysis to any of the invoked genes in Brugada syndrome is lacking.
The SCN5A gene is situated on chromosome 3p21 and encodes a large protein of 2016 amino acid residues [126]. Every α-subunit, which constitutes the major component of the cardiac Na+ channel complex, contains four homologous domains, each composed of six segments (S1–S6) and assembled with four ancillaries β-subunits (cytoskeleton proteins) to form the voltage-dependent cardiac sodium channel. The S5–S6 segments and the p-loop between them form the inner pore of the channel, which is high selective for Na+ ions. S4 segments act as the voltage sensor [127]. This channel belongs to a family with different isoforms and different biophysical properties according to its tissue distribution [8, 128]. In the heart it is responsible for the rapid initiating phase of the AP and thus plays an important role in impulse formation and propagation through the cardiac conduction system and muscle. The Na+ channel is dynamic and undergoes rapid structural transformations in response to the voltage changes across the sarcolemma. This process is known as “gating”. Upon membrane depolarization, the channel activates, allowing the opening of the pore. This increases channel permeability for Na+ ions. The resulting inward current causes the rapid upstroke of the AP. After a few milliseconds, fast inactivation of the channel occurs, a state in which the pore cannot re-open. Membrane repolarization is necessary to allow the Na+ channels to recover from inactivation into the resting state (closed state), from which they can re-open during the next cardiac cycle.
In the last years more than 200 SCN5A gene mutations (inherited Arrhythmia Database: http://www.fsm.it/cardmoc/) have been described in patients with the Brugada syndrome phenotype [29], alone or in combination with Long QT Syndrome type 3 (LQT3) and/or progressive cardiac conduction defects (PCCD, also known as Lev-Lenègre disease), AF, atrial standstill and dilated cardiomyophaty, diseases in which SCN5A mutations may also be present (Fig. 28.3) [8, 129–132]. Of interest, some SCN5A mutations may cause a combination of Brugada syndrome and LQT3 or Lev-Lenègre disease within the same family or even within the same individual [41, 133, 134]. While LQT3 associated SCN5A mutations generally increase INa, those associated with Lev-Lenègre disease reduce it, similar to those in Brugada syndrome [131, 135].
For a comprehensive compendium of SCN5A mutations in Brugada syndrome see the recent work of Kapplinger et al. [29].
Functional studies of mutant channels have been performed, up to now, with mutant proteins from at least 25 different SCN5A mutations. The common effect of SCN5A mutations associated with Brugada syndrome is reduction in sodium inward current (INa), during phase 0 of the cardiac AP, resulting from failure of expression of the mutant sodium channel in the cell membrane (trafficking) or changes in its functional properties (gating), resulting from: (1) shift in the voltage and time-dependence of INa activation and/or inactivation; (2) enhanced entry into an intermediate state of inactivation from which the channel recovers more slowly; (3) accelerated inactivation [30, 131, 136, 137].
The reduction in INa caused by the mutant sodium channels in Brugada syndrome is in agreement with the clinical observation that sodium channel blockers accentuate ST segment abnormalities in affected subjects [138]. Moreover, this finding concurs with the demonstration that Brugada syndrome patients who carry a SCN5A mutation have significantly more conduction disorders than non-carriers [110, 139, 140]. Also, within the cohort of the carriers, mutations which result in premature truncation of the sodium channel protein (insertions/deletions, nonsense and splice site mutations) are associated with longer PR and QRS intervals than missense mutations in which the mutant sodium channel is able to reach the membrane and is, at least in part, functionally active [31]. Despite the increasing number of SCN5A mutations recognized in Brugada syndrome, the proportion of clinically diagnosed Brugada syndrome probands who carry a SCN5A mutation is estimated around 14–22% [8, 55, 141]. Few other genes linked to the Brugada syndrome phenotype are now discovered. Still, other candidate genes other than SCN5A are unlikely to be major casual genes in this syndrome [142, 143]. In 2002, a novel mutation in the glycerol-3-phosphate dehydrogenase 1-like gene (GPD1L) on chromosome 3p22-24 has been described, linked to the Brugada syndrome phenotype in a large family [125]. This gene encodes a protein of 351 amino acids whose function in the heart still remains unknown, but the mutant protein, when studied in cell lines, was responsible for a diminished inward sodium current, similar to the other SCN5A mutations in Brugada syndrome studied so far [30, 42]. Mutations in GPD1L may also be responsible for sudden death in neonates [144]. More recently, genetic and heterologous expression studies revealed loss-of-function missense mutations in the gene which encodes the α1 subunit of the L-type calcium channel (CACNA1C) and its auxiliary subunits (CACNB2C and CACN2D1) in Brugada syndrome patients [36]. Several of these affected subjects also presented QT intervals shorter than normal [35]. Also, mutations in the β-subunits 1 (SCN1B) and 3 (SCN3B) of the sodium channel were found to have an effect on INa, leading to Brugada syndrome [37, 40]. Lately, also genes encoding for the transient outward potassium current, KCNE3 [145], KCND3 [39] and KCNE5 [38] were found to be associated with the Brugada syndrome phenotype. When the mutated KCNE3 was co-transfected with KCND3 in Chinese hamster ovary cells, this resulted in a significant augmentation in the amplitude of the Ito current, compared with the wild type [145]. Occasionally, mutations in the MOG1 gene, a gene encoding for a protein that regulates the expression of the sodium channel on the cell membrane, could be involved [146].
Other genes still await discovery. Good candidates appear to be, besides all genes that modulate ion current amplitudes during the initial parts of the cardiac AP, genes which encode adrenergic receptors, cholinergic receptors, ion-channel interacting proteins, transcriptional factors and transporters [147–149].
Though SCN5A mutations account for only 14–22% of all affected probands [26, 55], genetic testing is recommended during work-up in Brugada syndrome to support the clinical diagnosis, to identify affected relatives and to better elucidate the genotype-phenotype relationship in Brugada syndrome. Still, interpretation of genetic results must be performed with extreme caution and should be restricted to specialized centers where there is a structured collaboration between the cardiologists and the geneticist and where probands and his/her family members can be followed on a regular basis.
It has been shown in a Japanese cohort that the presence of a SCN5A mutation may somewhat influence the clinical course and VF recurrence [150]. Moreover, our group found that the type of the SCN5A mutation influences the severity of the clinical phenotype: carriers of a truncating mutation presented more severe conduction disorders than missense mutation carriers [31]. Interestingly, larger studies did not support the presence of a SCN5A mutation in the guidance of management of Brugada syndrome patients [10, 151, 152]. The explanation for this discrepancy could be due to the low prevalence of mutation carriers and the lack of functional data for many of the involved SCN5A mutants. Notably, the genetic background of an individual other than the primary SCN5A mutation, i.e. the presence of polymorphisms (either on the same or in other gene loci) [153] that can acts as strong modifiers, may also explain the differences in severity of phenotype. This concept is shown for the common polymorphism H558R [34, 154, 155]. Rare SCN5A variants are also described in healthy subjects [29]. Interestingly, common polymorphisms in the promoter region of SCN5A (Hap B) were also found to be associated with interindividual variability of expression and therefore variability in conduction parameters [156, 157]. These concepts add complexity in understanding and managing affected subjects and their family members. In daily practice, clinicians will deal with subjects carrying the primary mutation and suffering from severe arrhythmias and family members carrying the same genetic mutation that might not develop any arrhythmia. Even more strikingly, two groups showed that in several large SCN5A-related Brugada syndrome families, affected subjects (phenotype positive) did not carry the familial mutation (genotype negative) [43, 44], indicating that other factors may play a more important role than appreciated so far For all mentioned reasons, interpretation of genetic data in Brugada syndrome patients requires extreme caution [8].
ECG Characteristics
ST Segment Elevation Pattern
Typical electrocardiographic abnormalities have represented, since its first description, the fundamental aspect in recognition of subjects affected by Brugada syndrome [1, 2]. Particular attention was given to the presence of a (incomplete) right bundle branch block (RBBB), accompanied by ST segment elevation in the right precordial leads, not related to ischemia, electrolyte imbalance, or structural heart disease [4]. At present, diagnosis of Brugada syndrome revolves around characteristic ST segment elevations in leads V1–V3 or in leads positioned at more cranial intercostal spaces, whereas the presence of a RBBB is no longer required [23].
Three ECG repolarization patterns were described as potential manifestations of Brugada syndrome: type I ECG, referred to as coved-type, is the one illustrated in 1992 during the first description of the Brugada syndrome and consists of >2 mm J point elevation, followed by a down-sloping ST segment and a negative T wave; type II ECG, called saddle-back type, also shows a elevated J point (>2 mm) with a gradually descending ST segment that does not reach the baseline and gives rise to a positive or biphasic T wave; type III ECG could be of any of the previously described morphologies and is characterized by a smaller magnitude of ST segment elevation (≤1 mm) (Fig. 28.2). The presence of a type I ECG is required for the diagnosis [23]. Important considerations and cautions in interpretation of the ECG in diagnosing Brugada syndrome have to be taken into account. Firstly, the ST segment in Brugada syndrome is typically highly dynamic, exhibiting profound day-to-day variation in amplitude and morphology, even within the same patient [158–160]. It can even disappear temporarily. Clinical conditions of reduced cardiac excitability, like in the case of hyperkalaemia, can exaggerate ECG abnormalities in affected patients [161] and should be promptly recognized.
This aspect may contribute to possible bias and underestimation of the prevalence of Brugada syndrome. The presence of spontaneous ECG fluctuations measured on separate days in Brugada syndrome patients is associated with the highest risk of arrhythmic events and can be used as a non-invasive method for risk stratification, as well as the presence of late potentials on signal-averaged ECGs [162–164]. A spontaneous variation of ≥0.20 mV in right precordial leads ST segment elevation was found in affected patients who have had VF, but not in asymptomatic patients [165].
An inter-individual variation of the ST segment can also be observed between members of the same family who carry the same SCN5A mutation. The average magnitude of ST segment elevation does not differ, on average, between SCN5A mutation carriers and non-mutation carriers in Brugada syndrome [139]. In contrast, affected men have on average a higher degree of ST elevation than women [122]. Finally, many agents and conditions are reported to significantly influence ST segment elevation and therefore the risk of developing arrhythmias in Brugada syndrome. This phenomenon and its clinical implications are described further in this chapter.
Importance of Positioning of the Right Precordial Leads
The signature ST elevations in Brugada syndrome are usually observed in leads V1–V3, with rare occurrences in inferior or lateral limb leads [166–168]. More strikingly, leads positioned cranially from V1 and V2 in the third (V1IC3 and V2IC3) or second (V1IC2 and V2IC2) intercostal spaces often produce the most severe abnormalities, both in the presence and absence of pharmacological challenge [5, 169, 170], as also demonstrated with body surface mapping [3, 171]. This phenomenon emphasizes the crucial role of the RVOT area in the pathogenesis of Brugada syndrome [7]. The use of 87-lead body surface maps permitted to demonstrate that in 7/28 Brugada syndrome patients the typical ECG pattern was located at the level of the RVOT (second and third intercostals space), while conventional leads V1 and V2 registered only minimal ST segment elevation. Conversely, investigation of the more cranial leads in 40 control subjects did not reveal any significant ST elevation, neither at baseline, nor after disopyramide [3]. In 2006, we demonstrated that a more cranial positioning of the ECG leads increased the sensitivity of the flecainide testing in unmasking SCN5A-related Brugada syndrome [172]. These results were later on confirmed with the use of ajmaline in subjects with a family history of Brugada syndrome and/or SCD [173]. Although little is known about the specificity of the third intercostal leads and their prognostic information, we believe that ECG investigation in these more cranial leads should be performed whenever a case of Brugada syndrome is suspected [172, 174]. This topic should also be further addressed in a new Consensus Paper.
Other Electrocardiographic Parameters in Brugada Syndrome
Brugada syndrome has habitually been associated with the presence of right bundle brunch block, thought atypical because of the absence of a wide S wave in the left lateral leads [56]. Nowadays, the presence of a RBBB is no longer considered necessary for the diagnosis [23], though a widening of the QRS complex is frequently observed in patients affected by Brugada syndrome [172, 175]. Actually, signs of conduction defects are found at all levels (Fig. 28.4), particularly in patients carrying a SCN5A mutation [139, 140]: QRS axis deviation [1, 168, 176,177], P wave enlargement [110, 172], and PQ prolongation, presumably reflecting prolonged His-Ventricular conduction time [1, 56, 139, 178]. Moreover, sinus node dysfunction [177, 179, 180] and AV node dysfunction [139, 181, 182] have been extensively reported. In contrast, QTc duration generally is within the normal range [23, 56, 109], but it may be occasionally prolonged [1]. A new clinical entity was discovered in 2007 represented by coexistence of Brugada syndrome alterations and short QT interval, due to loss-of-function in calcium channels [35].
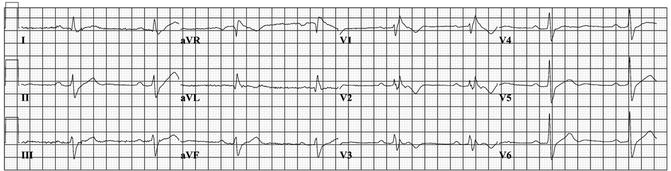
Figure 28–4.
Twelve lead ECG recorded at basal condition in a 49 year old male subject who suffered aborted sudden death. In addition to ST segment elevation in V1, signs of conduction disorders are present, including sinus bradycardia, PQ interval prolongation, left QRS axis deviation, and widening of QRS complex with a RBBB configuration, suggestive for Brugada syndrome
The presence of a SCN5A mutation significantly influenced the phenotype with more exhibition of clinically relevant conduction defects (first degree AV block, hemiblocks, complete RBBB, LBBB) in SCN5A carriers vs non carriers [139, 140], independently from the amount of ST segment elevation. Also, the type of SCN5A mutation influences the severity of conduction disorders in Brugada syndrome/PCCD patients [31]. Attention has also been paid to the recognition of other ECG criteria, in addition to the amount of J point elevation that may aid in identifying subjects at risk for sudden death. Five relatively new ECG parameters are: (1) S wave width in leads II and III, to be considered a mirror image of the electrical activity taking place in the RVOT [7]; these S waves were significantly wider in the individuals with a positive response to flecainide than in the negative responders [172]; (2) S wave width in lead V1 ≥ 0.08 s was shown to be a good predictor of arrhythmic events in Brugada syndrome patients [175] (3) baseline QRS width in lead V1 ≥ 110 ms significantly contributed to the prediction of a positive drug test [183]; (4) QRS width ≥120 ms on baseline ECG was associated with the occurrence of symptoms in a Japanese population [184]. The same cut-off value of prolongation of QRS duration on standard 12-leads ECG was already found to be a marker of arrhythmic risk and yielded a specificity and sensitivity of 70 and 52%, respectively, in identifying subjects with symptoms [185]; (5) fragmented QRS complex in V1–V3 or at the higher intercostal leads was found to be predictor of syncope in Brugada syndrome patients during 4 years follow-up [186].
Late Potentials
The presence of late potentials (LP) on signal-averaged ECGs (SAECG) has gained attention as a useful non-invasive method able to predict arrhythmic events in Brugada syndrome [17, 19]. LP are especially found in the anterior wall of the RVOT in symptomatic Brugada syndrome patients and can be exaggerated by infusion with flecainide [106]. They are generally regarded as delayed and disorganized ventricular activation (at the terminal portion of the QRS) and are related to a high risk of developing ventricular tachyarrhythmias [187]. The value of LP as predictors of arrhythmic events has been mainly tested in patients with structurally abnormal hearts, especially in studies of patients after a myocardial infarction [188]. In Brugada syndrome LP could represent delayed activation in the RVOT area [11], being in agreement with the depolarization disorder theory, or, according to some other authors, they may be an extension beyond the QRS of the second epicardial upstroke generated by a phase 2 reentry mechanism [12, 106].
Drug Tests
Pharmacological challenges utilize intravenous administration of sodium channel blockers, i.e., class IA (except quinidine) and IC, but not class IB [191] antiarrhythmic drugs. They are used to unmask concealed forms of Brugada syndrome because of their capacity to provoke/exaggerate ST segment changes [138, 182, 192–194]. Provocation tests are required when ST segment elevation is not initially present or when type II or III ECG patterns are seen [23]. However, provocation challenges are not recommended and could be even harmful in the presence of a spontaneous type I ECG [24, 195].
The diagnostic yield and safety of such tests, when performed with ajmaline or flecainide, have been recently reported in genotyped populations. Ajmaline (1 mg/kg body weight; 10 mg/min) was the most powerful drug [196], with a higher sensitivity (80%) and specificity (94%) [44] than flecainide (2 mg/kg; max 150 mg) (sensitivity 77%, specificity 80%) (Fig. 28.5) [172]. In Japan drug tests are performed with pilsicainide, a so-called pure sodium blocker (class IC). The protocol and the stopping criteria of the test with pilsicainide are similar to the European protocols [197]. Data on sensitivity and specificity of the tests with pilsicainide are missing, while ECG changes upon this drug (either in the positive or in the negative responders including investigation in the high precordial leads) and the safety issue have been carefully evaluated [197, 198]. Drug provocation tests with flecainide, ajmaline or pilsicainide have proven to be safe in large series of patients when conducted according to the guidelines of the European Society of Cardiology [23], since VT/VF did not occur [177, 197]. In particular, drug infusion must be discontinued when a type I ECG is reached or when PVCs/(non)sustained VT occur or when QRS duration increases by more than 30% of the basal value. If not discontinued, life-threatening ventricular tachyarrhythmias may develop (Fig. 28.4) [84, 195, 199]. Some authors suggested to abolish the stopping criterion of QRS prolongation during ajmaline administration because it may lead to an under recognition of positive cases [200]. Still, in their series, quite a high incidence of drug-induced arrhythmias was seen; moreover, more data is needed to evaluate whether such an aggressive protocol is leading to false positive results or not [201]. In patients with gross structural heart abnormalities, infusion with sodium channel blockers should also be avoided [202]. The presence of a SCN5A mutation seems to increase the risk of arrhythmias during infusion with sodium channel blockers [195].
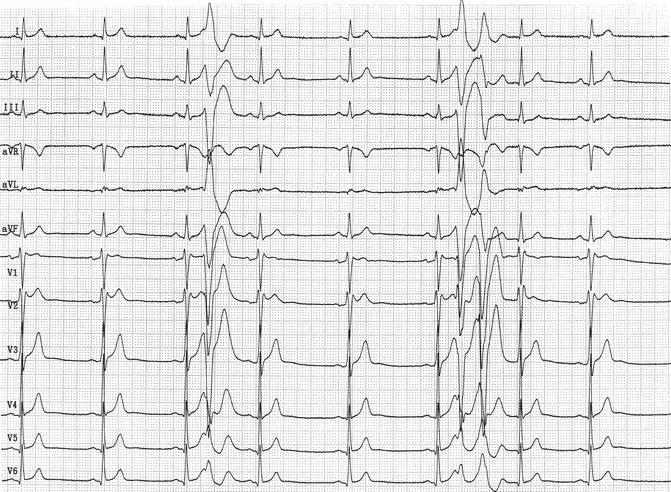
Figure 28–5.
ECG recorded after intravenous infusion of 80 mg flecainide in a 45 year old male subject showing a saddle-back ST segment elevation in leads V1 and V2 (type II) and the appearance of premature ventricular beats, isolated and in couples, from the right ventricular outflow tract. The ectopic beats show a short coupling interval. Flecainide challenge was performed to pose the diagnosis of Brugada syndrome after an aborted sudden death
Structural Abnormalities
One clinical characteristic of Brugada syndrome is the absence of gross structural abnormalities [1, 2]. Nonetheless, there is emerging evidence that Brugada syndrome may represent a mild form of right ventricle cardiomyopathy, not apparent with routine diagnostic tools [178, 203]. Similarities with arrhythmogenic right ventricular cardiomyopathy (ARVC) were pointed out especially by Italian researchers [178, 204] and were strengthened by the discovery of a SCN5A mutation in a family with ARVC [205]. On echocardiography, contraction of the RV was delayed by sodium channels blockade in Brugada patients compared with controls [16]. The sensitivity to detect slight structural abnormalities has become greater with electron beam CT scan and cardiac MRI. These methods have revealed RV wall motion abnormalities in at least three series of Brugada syndrome patients (these studies included control subjects) [206–209]. More recently, it has been shown that Brugada syndrome subjects show a significantly lower right ventricle ejection fraction on MRI then controls. Moreover, among affected subjects, the subgroup with spontaneous type I ECG also had significant RVOT enlargement and structural changes also of the left ventricle [210]. Notably, an Italian study had showed already in 2005 that in 4/18 Brugada syndrome patients who underwent biventricular endomyocardial biopsies, there were signs of right ventricular cardiomyopathy (n = 4), although the hearts appeared normal at non-invasive evaluation, including MRI [211] (no control group in this study). Interestingly, the presence of a SCN5A mutation was found in all of the four patients with cardiomyopathy-like changes on biopsy specimens. The remaining 14 heart specimens revealed changes compatible with myocarditis (n = 14). Furthermore, in 8/18 patients (45%), similar findings were also found in the left ventricle. Unlike this study, a German group found no evidence of inflammation or localized myocarditis in biopsies of Brugada syndrome patients. Still, they found fatty replacement in 4/21 patients in none of whom ARVC diagnostic criteria were fulfilled [212]. In addition, right ventricular fibrosis and epicardial fatty infiltration were documented in the explanted heart of a SCN5A mutation carrying young Brugada syndrome patient who experienced very many ICD discharges (129 appropriate shocks in 5 months) [21]. In this patient there were no clinically detected cardiac structural abnormalities, but it should be mentioned that MRI had not been performed due to ICD implantation, 10 years before cardiac transplantation. Fibrosis represents a substrate for conduction slowing, which is extensively found in Brugada syndrome. The amount of conduction disorders in affected patients is dynamic, as well as the degree of ST elevations, and is worse in the presence of type I ECG, compared with type II ECG or controls. The cell-to-cell transmission between myocardial cells seems to be impaired rather than the conduction times in the specialized conduction system [20]. These findings demonstrate a strong link between functional and structural abnormalities and also suggest the hypothesis that sodium channel mutations themselves are able to induce subtle structural derangements and myocardial cell death. This hypothesis has been elegantly tested in transgenic adult mice with SCN5A haploinsufficiency where a significant amount of cardiac fibrosis was found [213] and is supported by the clinical observation that certain SCN5A defects were associated with fibrosis in the conduction system and in the ventricular myocardium [214]. There is no end to complexity, since it is also acknowledged that in large Brugada syndrome families affected subjects might not even show the familial SCN5A mutation [43]. This work represents an eye-opener and leads to the hypothesis that genetic factors, other than alterations in the sodium channel gene, may also contribute to the occurrence of the disease.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


