Fig. 5.1
β-AR signaling. The major intracellular effect of the sympathetic transmitters norepinephrine and epinephrine is mediated by formation of 3’, 5’-cyclic monophosphate (cAMP), which increases the activity of the cAMP-dependent protein kinase A (PKA). PKA mediates a series of phosphorylations in diverse intracellular substrates, including the L-type Ca++ channels (LTTC), hyperpolarization-activated cyclic nucleotide-gated (HCN) channels, sarcoplasmic ryanodine receptors (RyR), phospholamban (PLN), myofibrillar proteins troponin I (TnI), cardiac myosin-binding protein C (MyBPC), and phospholemman (PLM). AC adenylyl cyclase, AR adrenergic receptor, ATP adenosine triphosphate, CNBD cyclic nucleotide-binding domain, G alpha-i and G alpha-s G protein alpha-subunit subtypes, SERCA sarcoendoplasmic reticulum (Adapted by permission from Triposkiadis et al. [21])
Furthermore, PK-A phosphorylates protein β-ARs themselves, resulting in partial uncoupling and desensitization of the receptor to further agonist stimulation [21]. As mentioned above, the β1– and β2-AR signaling pathways are both coupled to stimulation adenylyl cyclases. However, there is a marked difference between the two receptor subtypes. It seems that in human ventricular myocardium, stimulation of the β2 -AR is much more efficiently coupled to the production of cAMP than is the β1-AR [12, 22]. Myocardial contractility appears to be better associated with total β-AR density and β2-ARs do not appear to stimulate contractility more efficiently than β1-ARs [23].
Stimulation of β-ARs can also lead to the activation of MAP kinase members in a sub-selective way [24, 25], e.g. jun kinase and p38 kinase families [26]. These findings indicate that β-AR signaling pathways have significant mitogenic and proapoptotic actions. In particular, it has become clear that the β1– and β2-AR signaling pathways can be quite distinct [27]. For example, the β1-AR appears to be coupled primarily to the Gs/adenylyl cyclase/PK-A pathway and to modulation of L-type Ca++ channels and phospholamban. On the contrary, the β2-AR is not only coupled to Gs/adenylyl cyclase/PK-A, but also to the Gi pathway, and to “G protein-independent” functions such as regulation of the Na+/H+ exchanger.
As previously stated, cardiac function is mainly controlled by β-ARs and signaling and expression are tightly controlled by G protein–coupled receptor kinases (GRKs), which induce GPCR internalization and signal termination through phosphorylation. The GRKs are a family of cytosolic serine/threonine kinases consisting of seven isoforms that share structural and functional similarities [28]. The GRKs are classified into three subfamilies: (1) the rhodopsin kinase GRK1 and visual pigment kinase GRK7, (2) the β-AR kinases (or GRK2 and 3), and (3) the GRK4 group (GRK4–6). GRK2, GRK3, GRK5, and GRK6 are expressed in a wide variety of tissues, whereas other GRKs display a more restricted expression pattern. GRK2, initially identified as βARK1 (βAR kinase-1), and GRK5 are the most abundant GRKs expressed in the heart [29].
Cardiac contractility is mainly controlled by catecholamines that are either locally released from sympathetic neurons or by circulating catecholamines that are released from the adrenal gland. When catecholamine levels are increased, there is an acute physiologic stimulation of cardiac β-ARs resulting in an adaptive increase in cardiac output, which ultimately activates a negative feedback loop to suppress further catecholamine release. β-AR desensitization and signal cessation is mediated by GRKs via agonist-receptor interactions. GRKs phosphorylate β-ARs in an agonist-occupancy dependent manner. When β-ARs are stimulated, the dissociated G protein βγ subunits interact with GRKs, bringing the kinases within close proximity of the transmembrane β-ARs. The resultant phosphorylation of β-ARs by GRKs reduces the affinity of the receptor for the stimulatory G protein, Gs, and consequently increases the affinity of interaction for the inhibitory G protein, Gi [30]. Furthermore, phosphorylation leads to the interaction of β-ARs with β-arrestin adaptor proteins which facilitate the internalization of receptors into endosomic vesicles. These processes are involved in receptor resensitization/reexpression and in receptor downregulation.
5.3 β-ARs and Cardiovascular Diseases
Reduced β-AR density and activity associated with elevated cardiac GRK expression and activity have been described in various cardiovascular diseases (CVD). Clinical and experimental studies suggest that this GRK-mediated receptor desensitization is initially an adaptive/protective mechanism because chronic β-AR stimulation can be deleterious to the heart. In the failing heart, diminishing cardiac output triggers a vicious cycle of persistent sympathetic activation, resulting in further β-AR desensitization and maladaptive signaling due in large part to GRK-mediated receptor phosphorylation and loss of responsiveness to sympathetic stimulation. GRKs are upregulated in HF and thus contribute to the decrease in β-AR signal transduction [31]. Desensitization of the β-AR has been associated with upregulation of GRK2 both at the mRNA and at the protein level.
Failing hearts have been shown to have desensitized β-adrenergic receptor signaling and this adaptive mechanism may help maintain cardiac function in HF [32]. Norepinephrine stimulates all β-AR (β1-, β2– and β3-AR) subtypes and induces apoptosis (APO) in cardiomyocytes. Several mechanisms of β-AR stimulation-induced APO have been reported [33] (Fig. 5.2). However, not all subtypes of β-AR-mediated signaling induce cardiomyocyte APO. It is thought that the β1-AR-mediated pathway mainly contributes to this cell death process [34]. Although all three subtypes are coupled to Gsα, β2-AR and β3-AR are also linked to the Gi protein. The β2-AR exerts antiapoptotic effects through Gi βγ, phosphatidyl inositol-3 kinase (PI3K), and Akt (also known as protein kinase B) activation [35]. The β3-AR expression is upregulated in the failing heart [36]. It is reported, that unlike β1– and β2-AR stimulation by catecholamines, β3-AR negatively modulates ventricular contractility by activating endothelial nitric oxide synthase (eNOS). Catecholamines activate both positively inotropic and negatively inotropic pathways in human cardiomyocytes, with β1– and β2-AR mediating the positive inotropic effect, while β3-AR mediate a negative inotropic effect through eNOS overexpression [14]. Although the role of β3-AR-mediated signaling in cardiomyocyte APO is still unknown, it is possible that β3-AR exerts antiapoptotic effects through nitric oxide as well [37].
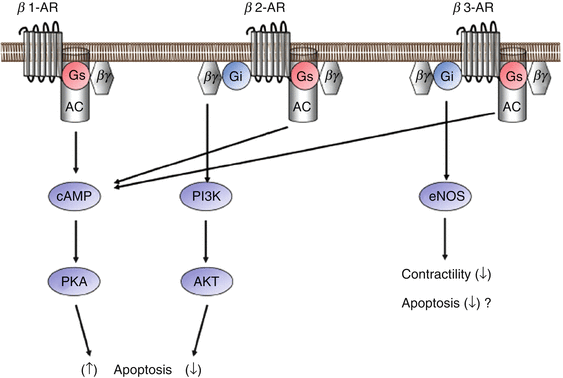
Fig. 5.2
The β-adrenergic receptor (β-AR) receptor-mediated signaling pathway for apoptosis. Three β-AR subtypes (β1-AR, β2-AR, and β3-AR) are expressed in cardiomyocytes. Although all 3 subtypes are coupled to Gs, β2-AR and β3-AR are also linked to the Gi protein. β1-AR is thought to be mainly involved in the apoptosis of cardiomyocytes. β2-AR exerts antiapoptotic effects through Giβγ, PI3K, and Akt activation. β3-AR negatively modulates ventricular contractility through endothelial nitric oxide synthase (eNOS) activation and might also be involved in the apoptosis of cardiomyocytes (Modified by permission from Fujita and Ishikawa [33])
The observation that stimulation of the β1-AR (Gsα coupling) appears to promote APO in cardiomyocytes whereas stimulation of the β2-AR (Gi βγ coupling) induces antiapoptotic effects [38] has been confirmed in animal models. In rats with myocardial infarction, treatment for 2 weeks with β2-AR agonists preserved cardiac contractility and reduced the number of apoptotic cardiomyocytes [39]. It has been shown that cardiac overexpression of the human β2-AR in a transgenic mouse [40] produced a significant increase in cardiac contractility while overexpression of a mini-peptide inhibitor of βARK produced a hyperdynamic mouse with no apparent histopathology [41]. In contrast, a transgenic mouse overexpressing the human β1-AR in ventricular myocardium [42, 43], unlike the β2-AR overexpressing mouse, demonstrated a time dependent reduction in myocardial contractility as well as marked myocyte hypertrophy, myofibrillar disarray and replacement fibrosis. In addition, the β1-AR transgenic mouse demonstrated upregulation of proapoptotic proteins and increased APO of cardiomyocytes. Thus regarding APO, the β1-AR mouse is more similar to transgenic mice overexpressing the stimulatory G protein, Gαs [44], than it is to the β2-AR mouse. It has been also shown that β-AR blockade in Gαs overexpressing mice can prevent the myocardial damage seen in this transgenic mouse model [45]. Therefore, attenuation of Gαs signaling, and presumably β1-AR signaling by β-blockade, strengthens the positive clinical data of the use of β-blockade in HF treatment.
5.4 Beta Adrenergic Receptors and Heart Failure
HF is associated to dramatic changes in neurohormonal balance. To compensate for the reduced effective arterial volume because of the diminished cardiac output, the autonomous nervous system (ANS) and the Renin-Angiotensin-Aldosterone System (RAAS) are highly activated. As a consequence of ANS hyperactivity, norepinephrine (NE) and epinephrine plasma levels increase, due to adrenal gland secretion and adrenergic terminal spillover, thus leading to chronic sympathetic stimulation of the heart. Such stimulation of cardiac β-ARs increases oxygen demand and myocardial work, thus contributing to cardiac muscle stress. Chronic ANS activation is also responsible for the selective down-regulation of β1-AR and for the functional uncoupling of both β1– and β2-ARs from their intracellular coupling mechanisms. These events are determined by activation of GRKs, particularly GRK2. Indeed, it has been demonstrated that, in chronic HF, GRK2 is up-regulated in cardiomyocytes, thus leading to a reduced responsiveness of the cardiac muscle to catecholamine stimulation [29]. GRK2-induced AR uncoupling and down-regulation also occur in the adrenal gland, where the inhibitory feedback on catecholamine release mediated by inhibitory α2-ARs is reduced, thus determining an increase of circulating epinephrine [46].
In the failing human heart, the β1-AR undergoes subtype selective downregulation at both the levels of mRNA and protein such that the β1:β2-AR subtype proportions become approximately equal [32]. Although there are some differences based on the etiology of HF, the extent of β1-AR downregulation correlates well with the severity of heart disease. The expression of myocardial β1-ARs also correlates well with systemic or coronary sinus plasma NE concentrations. A striking feature of HF is a characteristic set of molecular alterations in the components of the β-AR signaling pathway, including a decrease in β1-AR density and messenger ribonucleic acid, uncoupling of β1-AR from Gs, increased Gi protein and messenger ribonucleic acid, and impaired compartmentalization of cAMP/protein kinase A signaling [47–49]. β1-AR abnormalities have been attributed to the recruitment of both β-AR kinase-1 and phosphoinositide 3-kinase to the ligand-activated receptor complex [50, 51]. In addition, the levels of both Gi signaling proteins and the G-protein receptor kinases are increased. It seems that β-AR desensitization is a predominantly protective adaptation, which follows the increase in NE plasma levels that keeps intracellular cAMP concentration constant [52]. As mentioned above, this is further supported by the fact that overexpression of human β1-ARs in transgenic mice initially augments cardiac function but eventually leads to pathologic hypertrophy and HF [43, 53]. Many of the changes to the β-AR pathways associated with HF are also produced by the normal process of aging and there is also a good correlation between β1-AR density and age; the older the individual, the lower the β1-AR density [54].
Aging, like HF, does not appear to have a significant effect on β2-AR density. The role of β2-ARs in HF has not been delineated clearly [21]. In the failing human heart the β2-AR may be uncoupled from its signaling pathway but it is not downregulated. There are a number of potential explanations for this. First, the endogenous catecholamine, NE, is approximately 10–30-fold more selective for the β1– than the β2-AR. Second, the β1-AR appears to be localized to the synaptic cleft, thus it is more than likely exposed to higher concentrations of NE. Studies in transgenic animals have demonstrated that, in contrast to the early cardiomyopathy resulting from low-level (approx. 5-fold) cardiac overexpression of β1-ARs, even a 100-fold overexpression of β2-ARs in the mouse heart significantly increases cardiac contractile force without any cardiomyopathic consequences; extremely higher levels of overexpression (up to 350-fold) are needed to induce pathological changes [55]. Moreover, because β2-AR signaling contributes to an increase in the levels of Gi, it has been suggested that this may activate a protective antiapoptotic pathway in the setting of catecholamine excess. Indeed, in an experimental study, selective inhibition of Gi signaling in response to myocardial ischemia was associated with a marked enhancement of myocardial infarct size and apoptotic signaling [56]. Because of the relative preservation of β2-AR expression in the failing human heart, it has been the focus of therapeutic interest both for the development of β2-AR-selective pharmacological agents, both agonist and antagonist, and for a gene-therapy approach of increased cardiac contractility [57].
β3-AR mRNA is expressed in the human heart, but a corresponding receptor protein has not yet consistently been demonstrated. Furthermore, their physiological role remains highly controversial. For example, in human atria these receptors apparently do not promote cAMP formation and β3-AR have no effect on atrial contraction but, as stated above, in human ventricles β3-AR stimulation may exhibit negative inotropic effects [14]. However, more evidence is required that these effects indeed occur via β3-AR [58]. The role of β3-ARs in HF has not been fully elucidated. However, it has been proposed that in HF there is an excess of beta3-AR signaling, which exerts a negative inotropic effect potentially involving Pertussis toxin-sensitive G-proteins and activation of a NO synthase [36, 59].
In HF, sympathetic drive is selectively activated in several organ systems including the kidney, skeletal muscle and the heart. Increases in cardiac adrenergic activity result in marked increases in interstitial concentrations of NE within the failing human heart [60]. As discussed above, one consequence of the exposure of cardiomyocytes to high concentrations of NE is an alteration in cardiac adrenergic receptor pharmacology [48]. These changes decrease the sensitivity of cardiomyocytes to both endogenous and exogenous catecholamines. For example, there is a downward and rightward shift in dobutamine dose response curves in patients with heart failure compared to subjects with normal left ventricular function [61]. Maximal exercise workload as measured by peak VO2 also falls in proportion to the loss of cardiac β-receptors [62].
Cardiomyocyte growth is modulated in part by β1-, β2– and α1-ARs. Increases in signal transduction through these receptor pathways in the failing heart can contribute to pathological remodeling. Activation of these three receptors also results in increases in inotropic and chronotropic response. Myocardial energetics are adversely affected by these changes. Additionally, cardiac AR activation may promote arrhythmias. Another consequence of increased exposure of cardiomyocytes to increased concentrations of NE is myocyte toxicity. In tissue culture systems there is both a time- and concentration-dependent relationship between NE exposure and cardiomyocyte death [63]. The toxic effects of NE incubation can be partially blocked with the addition of a β-AR antagonist and completely prevented with the addition of both a β- and α-blocker to the culture media. Apoptosis of cardiomyocytes can be induced in tissue culture by NE. This effect appears to be mediated primarily through the β1-AR. Clinical examples of toxic effects of increased cardiac adrenergic drive include the observation of significant left ventricular dysfunction after catastrophic brain injury. However, the most compelling clinical support for the cardiotoxic role of cardiac NE are the beneficial effects that occur with the use of β-AR antagonists in patients with HF.
5.4.1 Beta Adrenergic Receptor Antagonists in Heart Failure
Beta adrenergic receptor antagonists (or β-blockers-BBs) are a wide and heterogeneous group of molecules acting as competitive and reversible antagonists of β-ARs. In addition to their ability to block β-AR-signaling, BBs show a variety of adjunctive actions that are often used as criteria for their classification. BBs can be classified according to β1-AR selectivity, α-AR antagonism and vasodilation, intrinsic sympathomimetic activity (ISA), pharmacokinetics and other properties like metabolic effects. BBs can be also classified into generations: (1) first generation, which are nonselective and competitively block both the β1– and β2-AR (propranolol, nadolol, timolol); (2) second generation, with much higher affinity for the β1– than for the β2-AR (atenolol, metoprolol, bisoprolol); and (3) third generation, which may be selective (celiprolol, nebivolol) or nonselective (bucindolol, carvedilol, labetalol) but all cause peripheral vasodilation mediated via α1-AR blockade (bucindolol, carvedilol, labetalol), β2-AR agonism (celiprolol), or nitric oxide synthesis (nebivolol) [64]. Cardioselectivity of β-blockers is dose-dependent and decreases with larger doses. Both selective and nonselective agents have negative chronotropic and inotropic effects. Selective agents have a less inhibitory effect on the β2– receptors and are less likely to cause peripheral vasoconstriction [65] and bronchial spasm. Exercise performance may be impaired to a lesser extent by β1-selective agents, partially because β2-blockade tends to blunt the exercise-induced increase in skeletal muscle blood flow. Finally, there are BBs that at low concentrations antagonize the cardiostimulant effects of catecholamines but at high concentrations cause cardiostimulation. These cardiostimulant BBs (e.g., pindolol, alprenolol, oxprenolol), widely known as nonconventional partial agonists, antagonize the effects of catecholamines through a high-affinity site (β1H-AR), but cause cardiostimulation mainly through a low-affinity site (β1L-AR) of the myocardial β1-AR [66]. Nonconventional partial agonists are considered potentially arrhythmogenic and should not be used for HF treatment.
The majority of BBs are partially or totally metabolized by CYP2D6, whose gene has more than 90 different variants [67]. Individuals homozygous for nonfunctional alleles are considered poor metabolizers. Several inconsistencies have been reported regarding the genotype phenotype correlations for intermediate or extensive metabolizers [68]. Poor metabolizers treated with metoprolol have a five-fold higher risk for adverse effects [69], whereas the adverse effects of carvedilol and bisoprolol are less influenced by the genetic background [70, 71]. Various protective mechanisms have been attributed to BBs, namely: (1) inhibition of catecholamine cardiotoxic effects; (2) β1-AR up-regulation (carvedilol is an exception); (3) attenuation of neurohumoral vasoconstrictive, growth-promoting, and pro-apoptotic systems (renin-angiotensin-aldosterone system, endothelin); (4) subendocardial coronary flow enhancement (as a result of diastolic prolongation); (5) restoration of the reflex control on the heart and circulation; and (6) improved myocardial performance by reducing heart rate and oxygen demand [72]. Among all BBs, bisoprolol, carvedilol, metoprolol and nebivolol are almost universally approved for the treatment of chronic HF [21]. Chronic BB therapy improves left ventricular performance and reverses left ventricular remodeling, reduces risk of hospitalization, and improves survival as will be further described.
5.4.1.1 Effects of β-Blocker Therapy in Heart Failure
Therapy of chronic HF with β-AR blockers has distinct acute (pharmacologic) and chronic (biologic) effects. The pharmacologic effects are the consequence of the acute changes in function that occur with withdrawal of sympathetic agonism and include a decrease in heart rate and contractile state. These detrimental effects can be tolerated only by initiating therapy with very low doses of β-blocker and slow up-titration of the dose over several weeks. There is significant variation in the severity of myocardial depression with different generations of BBs. Nonselective BBs such as propranolol reduce the contractile state and increase systemic resistance which profoundly decreases cardiac output [73]. As a consequence, the intolerance to initiation of propranolol is high (>20 %) and these agents have not been successfully used in long-term placebo-controlled trials. The initiation of β1-AR-selective β-blockers such as metoprolol and bisoprolol is much better tolerated by HF patients. This appears to occur because unblocked β2-ARs can support cardiac function either directly or indirectly (by β2-AR-mediated facilitation of sympathetic norepinephrine release). In addition, peripheral β2-ARs can mediate vasodilation. Thus, the overall detrimental effect upon organ perfusion is less severe than that observed with first generation compounds. β1-AR-selective BBs have been utilized successfully in prospective trials in HF with a tolerability >80 %. The third generation BBs possess vasodilator properties which counteract the negative properties of adrenergic withdrawal. This permits a more comprehensive blockade of cardiac adrenergic pathways with high tolerability (≥90 % in clinical trials of carvedilol and bucindolol).
5.4.1.2 β-Blockers Clinical Trials in Heart Failure
BB treatment in HF results in improvements in right heart hemodynamics, reversal of ventricular remodeling (as evidenced by reductions in left ventricular volumes, increases in right and left ventricular ejection fraction (LVEF), decreases in left ventricular mass and favorable effects upon left ventricular geometry), and improvements in HF symptoms [74]. To date, four specific BBs have been shown to be effective in the treatment of HF: metoprolol, bisoprolol, carvedilol and nebivolol [75]. (1) The Metoprolol in Dilated Cardiomyopathy trial, studied metoprolol tartrate (100–150 mg daily) which showed a trend toward a reduction in morbidity of patients with idiopathic dilated cardiomyopathy [76]. (2) The Metoprolol CR/XL Randomized Intervention Trial in Congestive Heart Failure (MERIT-HF) used a more effective formula with metoprolol succinate and showed a significant relative risk reduction in mortality in patients with New York Heart Association (NYHA) functional class II–IV [77, 78]. (3) The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II) demonstrated bisoprolol improved all-cause mortality by 32 %, sudden cardiac death by 45 % and reduced HF hospitalization by 30 % [79]. (4) The Australian New Zealand Heart Failure Research Collaborative Group (ANZ-HeFT) demonstrated significant improvement in left ventricular function and a significant reduction of left ventricular end diastolic volume index in a carvedilol group at 12 months in patients with NYHA class II or III [80]. Similarly, the Carvedilol Prospective Rando-mized Cumulative Survival (COPERNICUS) trial showed benefit in patients with NYHA class IV and an ejection fraction of <25 % with 38 % mortality risk reduction at 12 months [81]. The CAPRICORN trial focused on post myocardial infarction (MI) patients with LV dysfunction with ejection fraction of <40 % and incorporated current recommended therapy with the treatment of ACE inhibitors. The study demonstrated a 23 % further reduction of mortality with patients also receiving carvedilol 25 mg twice daily [82]. Long-term survival was good, particularly in patients with a nonischemic etiology for their dilated cardiomyopathy [83]. Finally, the SENIORS trial focused on elderly patients (>70 years old) with HF (ejection fraction <35 %) which demonstrated that nebivolol is an effective and well-tolerated treatment in this selected patient group [84].
5.4.1.3 Comparison Between β-Blockers
As previously mentioned, results from clinical trials demonstrated that blockade of β-AR signaling has beneficial effects in HF, both reducing mortality and improving symptoms of impaired cardiac function. Such protection has been demonstrated for different molecules belonging to BB class, and the degree of the beneficial effects was quite similar for any of the chosen drug. Thus, the idea existed that BB efficacy in HF was due to a “class effect.” On the other hand, many researchers have pointed out that specific pharmacodynamics and/or pharmacokinetic properties (β1-AR-selectivity, vasodilating actions, anti-oxidant activity, “pleiotropic effects”, adequate bioavailability and low drug-drug interaction risk) could represent valuable characteristics for preferring a specific β-blocker over others in the treatment of HF. To verify this hypothesis, a specific head-to-head clinical trial was conducted in 2003. The Carvedilol or Metoprolol European Trial (COMET) study aimed to compare the mortality risk-reducing effects in 3,029 patients with HF randomized to receive either carvedilol 25 mg twice daily or metoprolol tartrate 50 mg twice daily, after a mean follow-up of 58 months. The results demonstrated that carvedilol decreased mortality by 17 % with respect to metoprolol [85]. However, subsequent analyses showed that the degree of β-blockade reached with the formulation of metoprolol used in the study (metoprolol tartrate) was not adequate, and probably not similar to that of the carvedilol arm. Basically, metoprolol tartrate should have been titrated up in the COMET study [86]. An answer to this issue has been also pursued by several meta analyses, most of which have been focused on carvedilol, possibly because of its unique pharmacodynamics profile. Nevertheless, results from these studies seem to be conflicting. In a systematic review of 11 randomized controlled trials in 5,207 patients, it was found that carvedilol reduced all-cause mortality in patients with HF significantly more when compared to other β-blockers such as atenolol, bisoprolol and metoprolol. The superiority of carvedilol over other β-blockers could be attributed to the special properties of carvedilol, such as: antiarrhythmic effect (and consequent reduction in the risk of sudden death), a more sustained increase in LVEF when compared to other β-blockers, blockade of up-regulated β2-AR and vasodilation [87]. By contrast, in another meta-analysis of 21 trials including 23,122 HF patients, no significant differences were found in mortality outcome among the β-blockers considered (atenolol, bisoprolol, bucindolol, carvedilol, metoprolol, and nebivolol), thus concluding that the beneficial of β-blockers were due to a “class effect.” Nonetheless, carvedilol showed the lowest cardiac mortality among all β-blockers tested, although this was not statistically significant [88]. Lack of clear data from clinical trials regarding the superiority of a given BB is reflected in the 2012 ESC guidelines for the management of HF, in which the use of a particular BB over the others is not recommended [75].
5.4.1.4 Pharmacogenomics and Therapeutic Impact of β-AR Polymorphisms
As for many other pharmacological treatments, variability in clinical and functional outcomes exists also for BB administration in HF patients. Actually, BB treatment fails to improve LVEF in a variable proportion of subjects, while a minority of patients experience worsening of HF symptoms during BB titration [89]. Such differences could be explained, at least in part, by genetic variation influencing BB pharmacodynamics and/or pharmacokinetics. To date, polymorphisms in β1-AR, β2-AR, GRK-5, CYP2C6, NET and UGT1A1 have been associated to variability in BB response.
Many studies have examined the impact of the aforementioned polymorphisms in response to β-blockade among HF patients. Preliminary studies in patients treated with various BBs have demonstrated a survival benefit [90] and a significant reduction in left ventricular end diastolic diameter [91] for those who carried the Gly49 allele as compared to the Ser49 homozygous patients. A similar reversal of the remodeling process has been reported for Arg389-homozygous HF patients treated with carvedilol as compared with Gly389-homozygous [92] and Arg389Gly-heterozygous patients [93]. However, neutral results have also been reported. For example, no survival benefit was observed for the Ser49Gly or the Arg389Gly patients treated with carvedilol [94, 95], bisoprolol [94], or metoprolol [95], and for the Arg389-homozygous or Gly389 patients treated with metoprolol CR/XL [96]. Finally, a dose-dependent response to beta-blockade has been reported, whereby low doses in patients carrying the Gly49 allele portend worse outcomes than in Ser49-homozygous patients, whereas in higher doses, genotype dependent differences are not observed [97]. More recent data, however, suggest improvement in the therapeutic effect of bucindolol by pharmacogenetic targeting. In the deoxyribonucleic acid substudy of the Beta Blocker Evaluation of Survival Trial (BEST) [98], patients carrying the Arg389 genotype had a greater agonist promoted ventricular contractility along with improved age-, sex-, and race-adjusted survival than the Gly389 carriers [99].
Taken together, these results, although suggestive of the possibility to select BB according to a particular genotype, are not conclusive, thus requiring large prospective clinical trials. In this respect, the use of genetic polymorphisms as potential tools to guide therapeutic strategies seems a promising new approach. The subject of genetic polymorphisms is described in Chap. 7.
5.4.1.5 GRK2 as a Potential Target in Heart Failure Treatment
As mentioned above, the up-regulation of GRK2 (and also probably GRK5) caused by SNS activation acts as a “brake” for cardiac β-AR signaling and this is a partial reason why BBs have beneficial effects in HF as they can block the noxious effects of catecholamines on the cardiomyocyte. However, increased GRK2 activity has proven to be maladaptive in HF and its inhibition has emerged as a therapeutic target [100]. Interestingly, chronic BB use in HF models has been shown to decrease GRK2 expression levels, which could contribute to BBs’ therapeutic effects [101, 102].
It has been shown that lowering GRK2 expression or activity in the injured, stressed or failing heart can prevent or reverse ventricular dysfunction at the functional and morphological level [103]. This has been shown using a peptide inhibitor known as the βARKct (Gβγ protein sequestering peptide) and also in GRK2 knockout (KO) mice. First, cardiac-specific βARKct transgenic mice were created showing increased inotropic reserve [104] and these mice have been used to prevent HF in several genetic mouse models [105–107]. In addition, viral-mediated βARKct delivery to rats, rabbits, and pigs have shown significant beneficial effects including improved cardiac function and reverse ventricular remodeling [102, 108–111]. Similar findings were noted by induced KO of GRK2 in mice, after HF was evident, and this is consistent with βARKct-mediated inhibition of GRK2 [112]. Although there are some minor differences between GRK2 inhibition with βARKct and GRK2 KO, the data is overwhelming in supporting GRK2 inhibition as beneficial in the failing heart [112–114]. Importantly, a large animal HF study has been done that demonstrates the clinical potential of βARKct-mediated gene therapy [111]. This study used adeno-associated virus serotype-6 (AAV6) to deliver βARKct to a post-ischemic HF model in the pig [111, 115]. It was found that AAV6-βARKct delivery ameliorated LV function, and suppressed adverse cardiac remodeling and fetal gene expression in this model [111]. Of note, this study in pigs had similar results found with AAV6-βARKct delivery to a rat model of HF where it was also shown that βARKct worked significantly better that β-blockade alone and the two were complementary together [102]. Of importance, both of these studies showed that chronic GRK2 inhibition results in significant lowering of catecholamines and aldosterone demonstrating feedback to decrease the neurohormonal outflow associated with negative prognosis in HF [102, 111]. These two studies in particular are important as they show reversal of the disease process and the pig study closely reflect human pathophysiology and is a pre-requisite for future clinical trials.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


