Fig. 1.1
Schematic illustration of monogenetic and multifactorial cardiovascular diseases. In monogenetic diseases (left), genetic predisposition is responsible for disease onset induced by a relatively clear molecular pathway. Outcome and prognosis may then be alleviated or worsened by complex modifications of environmental factors. Multifactorial cardiovascular diseases are triggered by an imbalance between environmental (G×E) and genetic factors (G×G). The molecular pathways of the diseases may then again be modulated by heritable or environmental modifications of elements within the underlying molecular pathways
1.1.1 “Mendelian” and Monogenic Diseases
If the occurrence of a disease in a familiar anamnesis follows a clear Mendelian autosomal-dominant, autosomal-recessive, or gonosomal (X, Y) pattern, it is highly unlikely that multiple independent gene defects cause the disorder in a linked way. Therefore, these diseases are also termed monogenic diseases, as they can be convincingly traced back to single genes. This simple concept of monogenetic diseases has in the last decade been challenged by reports of incomplete penetrance as well as the identification of triallelic forms of inheritance (Eichers et al. 2004), where mutations in a small number of genes interact genetically to manifest a certain phenotype. A monogenic pathophysiology, however, is generally characterized by a relatively clear causal pathway that is a sufficient and, in most cases, inevitable cause of disease. It has been observed in clinical practice that the impact of at least some of these diseases may, nevertheless, be alleviated or worsened by modifications, often age related, of the affected pathway (Miyamoto et al. 2012). Accordingly, environmental factors like smoking or physical activity may have a significant impact on the prognosis of monogenic diseases. Considering the number of possible modifications, the complexity of involved molecular pathways that have been identified in some monogenetic diseases may even approximate those of typical multifactorial disorders.
The prevalence of monogenetic diseases is typically very low and estimated to range from 0.005 % in the rarest forms up to 0.1–0.2 %, as observed in hypertrophic cardiomyopathy (HCM), the most common cardiac disease with Mendelian inheritance. More than 50 % of all cases of HCM occur familially, following an autosomal-dominant pattern, and 22 % of the first-degree relatives in these families were shown to be affected (Greaves et al. 1987). Despite the relatively clear causal origin, however, the global view on all possible forms of HCM reveals a genetically highly complex situation of pathogenesis, because mutations in several different molecular targets are linked to HCM. While more than 80 % of the mutations involved affect the myosin heavy chain gene (MYH7), troponin T (TNNT2), and myosin-binding protein C (MYBPC3), the HCM phenotype, as characterized by a left ventricular myocardial hypertrophy, may also be caused by mutations affecting a number of other sarcomeric proteins, including troponin I (TNNTI2), α-tropomyosin (TPM1), essential and regulatory myosin light chain (MYL3, MYL2), titin (TTN), β-myosin heavy chain (MYH6), and muscle LIM protein (MLP). These mutations of other sarcomeric proteins have been demonstrated to account for 30–60 % of the sporadic forms of HCM (Richard et al. 2003). In addition to about 300 identified mutations of sarcomeric proteins, mutations of Z-disc proteins have also been found to be involved in HCM. Here LIM domain binding 3 (LDB3) and muscle LIM protein (CSRP3) as well as vinculin and α-actinin 2 were demonstrated to be affected (Gehmlich et al. 2004; Vasile et al. 2006; Wang et al. 2010a). HCM is, therefore, considered a genetically heterogenic disease, as also indicated by the low likeliness of two nonrelative HCM patients to carry the same mutations. An estimated 3 % of affected individuals carry more than one mutation (Van Driest et al. 2004).
Despite the diversity of genetic backgrounds, the resulting molecular pathways leading to HCM involve different elements of a common functional unit in the myocyte. The myosin heavy chain protein forms the thick filament of the sarcomere, while the thin filament is comprised of actin, α-tropomyosin, and the troponins. These structures are stabilized by myosin-binding protein C and titin. A calcium-triggered signal then leads to an ATP-dependent conformational change in the myosin heavy chain, resulting in filament sliding and muscle contraction. Mutations in sarcomeric genes are the sufficient and the primary cause of HCM as demonstrated by studies using transgenic animals in genetically engineered mice and rabbits expressing human HCM mutations that induced the development of histopathological phenotypes comparable to human HCM (Marian et al. 1999; Oberst et al. 1998; Vikstrom et al. 1995). Analysis of protein structure/function relationships of the mutated human β-myosin heavy chain and the broad local distribution of HCM mutations across the protein suggests that the HCM mutations are not necessarily affecting the basic functions of the protein, like ATP hydrolysis or actin binding, but may relate to interaction with other proteins of the sarcomere (Rayment et al. 1995). For some affected sarcomeric proteins, clear mutation-induced functional consequences have been demonstrated. This includes disturbed intrinsic ATPase activity of α-cardiac muscle actin (Muller et al. 2012) or alterations in the concerted conformational change of the α-tropomyosin/actin complex (Rysev et al. 2012). The common result of most sarcomeric mutations involved in HCM appears to be a change in myocyte contractility.
The chain of events leading to left ventricular hypertrophy is still unclear. One proposed mechanism is decreased contractility of cardiac myocytes, leading to cellular stress and production of trophic factors (Marian 2000), which ultimately causes transcriptional reprogramming and hypertrophy. Another hypothesis suggests that abnormal force development of the myocytes triggers a reflectory increase in sympathetic activity, possibly due to changes in diastolic filling (Ostman-Smith and Wettrell 2000). Because both of these hypotheses are supported by several studies (Fineschi et al. 2005; Lim et al. 2001; Pace et al. 2004; Patel et al. 2000), a concerted contribution by these mechanisms might be considered. The resulting left ventricular hypertrophy and fibrosis are typically associated with enhanced systolic function and impaired diastolic function. Depending on the genetic base, the clinical spectrum of HCM is wide ranging, spreading from relatively benign forms to heart failure and sudden cardiac death.
Recent progress in genetics, including the completion of the human genome project, has enabled the identification of a number of cardiovascular target proteins and helped to identify monogenic forms of common disorders. One example is coronary heart disease, a disorder typically displaying multifactorial causality. While common forms are mainly influenced by classical risk factors like dyslipidemia or diabetes mellitus, monogenetic forms have been traced back to LRP6, a protein involved in the Wnt-signaling pathway (Mani et al. 2007), and a less clear causality has been demonstrated for MEF2A, a transcription factor located in the nucleus of proliferating smooth muscle cells (Lieb et al. 2008; Weng et al. 2005). Monogenetic forms were also found in arterial hypertension and atrial fibrillation (see Chap. 4). In arterial hypertension, mutations of the mineralocorticoid receptor cause a partial constitutive activity of the receptor, leading to hypertensive crisis in female members of affected family trees. Inherited mutations in renal ion transporter systems can also cause hypertension, as can be seen in a form of hyperaldosteronism, where a crossing-over induced hybridization of aldosterone synthase and 11β-hydroxylase forms a protein that is no longer under control of by angiotensin II but activated by adrenocorticotropic hormone (ACTH) (Lifton et al. 1992).
1.1.2 Multifactorial Cardiovascular Disorders
Most CVDs can be triggered by genetic as well as environmental factors, which are typically able to induce a disease in patients with genetic predisposition, i.e., who are carrying common variants in susceptibility genes. Environmental factors are characterized, in principle, as avoidable external modulators of cellular pathways and comprise a wide range of physical, chemical, biological, dietary, and psychological factors, including radiation, toxins, pathogens, malnutrition, physical strain, and mental stress.
Most multifactorial diseases are based on complex molecular pathways, making it difficult to distinguish between causation and correlation within the range of disease-promoting factors. The situation is even more complex, because recent scientific advances indicate that genetic and environmental factors are often linked and that different pathological gene expression patterns may interact with other genes as well as environmental factors in a synergistic or antagonistic way. Young scientific disciplines like environmental genomics and epigenetics are positioned to shed light onto these topics using multidisciplinary approaches.
Recently, a database for gene-environment interactions (G×E) of the human genome compiled from literature was presented (Lee et al. 2011), showing 554 significant interactions that were associated with obesity, metabolic disorder, and CVDs. An example of such a gene-environment interaction is provided by a study (Romanoski et al. 2010) demonstrating a significant interaction between genotypes of explanted human endothelial cells and their exposure to oxidized phospholipids, a common scenario found in atherosclerosis. About one third of 59 of the most regulated gene transcripts showed evidence for gene-environment interactions, and a direct local interaction was observed for FGD6, a protein that is hypothesized to represent a Rho family guanine nucleotide exchange factor (RhoGEF) and catalyzing GDP/GTP exchange for small G proteins involved in cytoskeletal pathways.
Another well-established example demonstrating modification of gene expression pattern by environmental factors is found in nonphysiological hypertrophy of the heart. At birth, metabolism of the heart switches from the hypoxic “intrauterine mode” to the oxidation of fatty acids, and a mature set of enzyme proteins is expressed. The fetal metabolic program, however, can become reactivated as soon as the heart is stressed by hypoxia, ischemia, atrophy, or hypertrophy (Kuwahara et al. 2012). This adaptive process of re-expression of fetal genes is seen as a hallmark of pathophysiological hypertrophy and demonstrates the direct effect of environmental conditions on genetic expression (Kehat and Molkentin 2010).
Interactions among genes are defined as phenotypic effects of two or more coincident gene modifications that differ from those observed for independent contributions of each gene. Because of their relatively high abundance within populations, many studies investigating these synergistic effects among genes refer to polymorphisms. Polymorphisms are described as variance among genes with a relatively high frequency of occurrence >1 % that cannot be explained by accident mutations but are rather due to general variability induced by allelic drift, i.e., the frequency of occurrence of an allele, and that are stabilized by evolutionary selection. A large number of polymorphisms have been identified as risk factors in various CVDs (see Chap. 4).
Interactions between different genes (G×G) relevant for cardiovascular pathophysiology have been described in several studies. One example is given by a population-based study, indicating that genes involved in the renin-angiotensin, bradykinin, and fibrinolytic systems synergistically modulate plasma concentrations of plasminogen activator and plasminogen activator inhibitor type 1 (Bentley et al. 2010). Here it is demonstrated that the coincidence of polymorphisms may generate a much higher CVD risk than the simple arithmetic sum of the relative risk factors, as derived from single polymorphisms.
1.2 Common Molecular Pathways in Cardiovascular Disease
In multifactorial CVDs, gene-gene interactions and gene-environment interactions are linked to a plethora of molecular pathways that result in specific cellular dysfunctions, morphological changes, or cell death. The number of pathways that have been identified so far correlates with the diversity of CVDs and, therefore, does not allow for a comprehensive presentation here. However, a selection of molecular mechanisms will be presented within the next paragraphs. Recent scientific advances have helped to identify numerous molecular pathways linked with the pathogenesis of CVDs. It should be kept in mind that most of the results, however, especially those aiming to elucidate molecular pathways, were obtained by using animal or even cellular model systems.
Some identified pathways are involved in several forms of CVDs and may thus be considered as pivotal molecular mechanisms, even if their role may be emphasized in a certain pathophysiological context. The common pathological key events represented by these pathways are, in a broader sense, posttranslational modifications of proteins and other molecules, including oxidation, phosphorylation, and glycation processes.
1.2.1 Oxidative Stress
Reactive oxygen species (ROS) are characterized by unpaired valence electrons in the outer shells of atoms and thereby able to react with a large number of molecular target proteins, lipids, carbohydrates, and nucleic acids. Consequently, ROS have been implicated in many diseases as well as aging-related pathophysiology of the cardiovascular system (Chen et al. 2012).
ROS levels in cells increase after exposure to environmental factors like UV light, heat, or ionizing radiation and can also be produced by metabolism of xenobiotics. However, ROS are also generated by physiological processes, including activity of the mitochondrial respiratory chain and enzymatic reactions as catalyzed by uncoupled eNOS and NADPH oxidases, lipoxygenase, monoamine oxidase, and xanthine oxidase. This general oxidative stress is counterbalanced by the activity of a number of cellular antioxidant enzymes, including glutathione peroxidase (GPx), catalase, and superoxide dismutase (SOD), as well as some exogenously obtained dietary compounds (Shao et al. 2012).
The most common radicals are superoxide anion radical (•O2 −), hydrogen peroxide (H2O2), hydroxyl radicals (•OH), NO radicals (•NO), nitrogen dioxide radicals (NO2), peroxynitrite (ONOO−), and hypochloride (ClO−).
The reaction of the superoxide anion, with the main endothelial relaxing factor NO, is extremely efficient. Consequently, one obvious cardiovascular outcome of enhanced superoxide production is decreased NO bioavailability. The reaction product of •O2 − and NO, peroxynitrite, is known to induce the activity of poly ADP-ribose polymerase (PARP), an enzyme that has been shown to induce cellular dysfunction and metabolic disorders in cells and has been demonstrated to be involved in vascular aging (Burkle et al. 2005; Csiszar et al. 2005).
Production of ROS in the cardiovascular system seems to be tightly linked to the renin-angiotensin system (RAS), because chronic inhibition of the RAS or genetic deletion of the angiotensin II type 1 receptor suppresses peroxynitrite formation during aging. Angiotensin II has been identified as a major stimulus for NAPDH oxidase and, thus, a key element for accumulating oxidative stress during aging (Oudit et al. 2007; Wang et al. 2010b).
Oxidative stress is well known to be involved in initiation and progression of the pathogenesis of atherosclerosis. Atherosclerosis and its clinical representations are directly linked to oxidation of lipids bound to LDLs that accumulate in the network of the extracellular matrix of the subendothelial space. While immobilized, ROS secreted from several pathways of the vessel wall can reach the LDL and induce lipid oxidation (Witztum and Steinberg 1991). LDL oxidation is seen as a two-stage process with an initial, relatively slow oxidation, characterized by only small changes in ApoB and a more rapid second stage initiated by arrival of monocytes, which provides a strong additional source of ROS when converted to macrophages. This oxidative potential of monocytes/macrophages may also contribute to the pathophysiology of hypertension (Lim et al. 2006) and is mainly due to the physiological accumulation of ROS in the phagosome by NOX2 activity (Judkins et al. 2010). This accumulating oxidative stress leads to modification of the LDL receptor and loss of its ability to recognize cholesterol. As a consequence, LDL is transported by the receptors without limiting the cholesterol uptake, resulting in cellular cholesterol accumulation and formation of foam cells (McLaren et al. 2011).
Epidemiological Examples Demonstrating Relevance of Cellular Redox Balance
A lectin-like 52-kD receptor for oxidized LDL (LOX-1) is a scavenger receptor present primarily on vascular endothelial cells and binds and regulates oxidized LDL. Ligand activity of LOX-1 has been found associated with non-HDL cholesterol, triglycerides, history of smoking, and risk of coronary artery disease (Inoue et al. 2010; Uchida et al. 2011).
Oxidation of LDL has been demonstrated to promote atherosclerotic inflammation by affecting monocyte tethering, activation and attachment (McEver 1992). P-selectin, a tethering protein, has been demonstrated to be upregulated and released from endothelial cells in the presence of oxidized LDL (Vora et al. 1997). In addition, oxidized LDL in endothelial cells was shown to induce several pro-inflammatory factors including monocyte chemoattractant protein 1 (MCP-1) (Cushing et al. 1990), monocyte colony-stimulating factor (M-CSF) (Rajavashisth et al. 1990), and human growth-related oncogene (GRO) (Schwartz et al. 1994). Thus, initially limited oxidation of LDL may lead, via a positive feedback mechanism, to accumulated production of ROS. Further development of the atherosclerotic lesion is promoted by activation of transcriptional programs that trigger an inflammatory response and induce arterial calcification that alters the mechanical properties of the vessel wall, enables infiltration by monocytes, and, in advanced stages, triggers plaque rupture.
Recent findings suggest that ROS-generating pathways are modulated by Ca2+, and a crosstalk of signaling between ROS and Ca2+ has been demonstrated for several systems (Lee et al. 2010; Sun et al. 2011). While Cav1.2 and other plasmalemmal calcium-conducting channels have been shown to be regulated by ROS (Amberg et al. 2010; Cioffi 2011; Florea and Blatter 2008; Hool et al. 2005; Poteser et al. 2006; Weissmann et al. 2012), intracellular Ca2+ stores may be compromised by expressional downregulation of calsequestrin (Hanninen et al. 2010) or ROS-induced modification of the cardiac ryanodine receptor (Donoso et al. 2011). In turn, ROS-generating NADPH oxidase activity (Nox5) was shown to be regulated by intracellular Ca2+ levels via CAMKII (El Jamali et al. 2008; Pandey et al. 2011) and •O2 − production in mitochondria is closely related to Ca2+ concentrations (Victor et al. 2009), enabling feed-forward interactions that may finally lead to cellular Ca2+ overload (Peng and Jou 2010).
1.2.2 Kinase Pathways
Important cellular functions such as growth and differentiation are governed by environmental and genetic factors that alter signal transduction pathways culminating in altered gene expression. Critical components of many of those pathways are protein kinases, which phosphorylate and regulate cellular substrates including transcription factors. Kinase pathways are characterized by a cascade-like chain of reactions suitable to potentiate even very weak initial events into robust cellular signals.
A specific set of serine/threonine kinases is activated by factors induced via environmental disturbances like heat, inflammatory signals, ischemia, and genotoxic stress. These kinases have therefore been termed stress-activated protein kinases (SAPKs), also known as c-Jun N-terminal kinases (JNK). These SAPKs, together with kinase p38, represent a common element in the pathophysiology of many CVDs (Bogoyevitch et al. 1996; Choukroun et al. 1998; Johnson and Nakamura 2007; Liang and Molkentin 2003; Seko et al. 1997).
A molecular mechanism involving SAPKs is part of the pathophysiology of ischemic-reperfusion injury (Clerk et al. 1998). Ischemic-reperfusion injury is characterized as initial anoxic cell injury, triggering decreased mitochondrial ATP production, activation of hydrolases, and compromised membrane integrity. Resupply of blood in the second phase is then followed by inflammatory cellular responses. Reperfusion of ischemic tissue can activate genetic programs, which induce cells to dedifferentiate, proliferate, or undergo apoptosis. While the exact initial events of the pathway are still unclear, the radical scavenger GSH has been shown to prevent activation of SAPKs, substantiating a key role of oxidative stress. SAPKs, however, are the predominant ATF-2 (activating transcription factor 2) kinases activated by reperfusion (Pombo et al. 1994). After ischemia and reperfusion, SAPKs target transcription factors of multiple promoters, hypothesized to be c-Jun/ATF-2 heterodimers, providing a potent mechanism to induce a large number of genes. Their targets are the ATF/CRE and jun2 TRE motifs of the c-jun promoter (Morooka et al. 1995). The activated genes typically encode for proteins involved in apoptotic processes, inflammatory pathways, and heat shock proteins (Conti et al. 2007).
SAPKs were shown to be involved in inflammatory responses of macrophages and smooth muscle cells of the artherosclerotic plaque (Metzler et al. 2000). Inhibition of p38 prevents the cumulative production of cytokines like IL-1β and TNF-α by interruption of feed-forward amplification on a translational level (Feng et al. 2001).
5′ AMP-activated protein kinase (AMPK) is an enzyme that regulates metabolic functions of heart, as well as liver, brain, and skeletal muscle, and helps to normalize cellular lipid, glucose, and energy imbalances. AMPK is thus seen as an interesting drug target for the treatment of metabolic syndrome and was shown to regulate the function of lipoprotein lipase (LPL) (An et al. 2005), an enzyme that is critical for the fatty acid supply of the cardiomyocyte and the GLUT4 glucose transporter in the heart. AMPK has also been reported to play a role in ion channel activation (Alesutan et al. 2011; Ikematsu et al. 2011), ischemia-reperfusion injury (Kambara et al. 2012; Omar et al. 2012; Paiva et al. 2011), oxidative stress (Colombo and Moncada 2009), hypertrophy (Jiang et al. 2010), and other pathophysiological pathways (Beauloye et al. 2011; Heidrich et al. 2010).
Another group of kinases that has been identified to be critically involved in CVDs including angina, cerebral ischemia, myocardial ischemia, and cardiac hypertrophy is the group of Rho kinases. Rho-associated coil-forming protein kinases (ROCKs) are downstream targets of RhoA, a GTP-triggered molecular switch representing a key element in the regulation of some basic cellular functions like contractility, migration, proliferation, and apoptosis.
Epidemiological Examples Demonstrating the Relevance of Cellular Kinase Pathways
An evidence of involvement of protein phosphorylation in cardiovascular disease, activity of Rho-associated kinases (ROCK) in circulating leukocytes has been proposed to be a marker of cardiovascular risk. ROCK activity reportedly showed significant positive correlations with flow-mediated vasodilation (FMD), reflecting endothelial function, as well as with various cardiovascular risk factors, such as body mass index (BMI), systolic blood pressure, LDL cholesterol, and Framingham risk factor score, a cumulative cardiovascular risk factor (Hidaka et al. 2010; Soga et al. 2011). Moreover, smokers and patients with vasospastic angina have been reported to show increased ROCK activity (Hidaka et al. 2010; Kikuchi et al. 2011).
Typical target proteins of ROCKs are regulatory elements of the cytoskeleton, adducin (Fukata et al. 1999), MARCKS (Mueller et al. 2005), intermediate filaments like desmin and vimentin (Inada et al. 1999), as well as proteins of contractile machinery like calponin (Qu et al. 2008), troponin (Vahebi et al. 2005) and PTEN (Chang et al. 2006). Activation of ROCKs has been demonstrated to regulate the Ca2+ sensitivity of the contractile proteins by phosphorylation of MYPT-1, a regulatory subunit of MLC phosphatase, and this modulation is of special importance in contractile cells (Ohtsu et al. 2005).
In addition, gene expression in vascular smooth muscle cells has been demonstrated to depend on this group of kinases. Myocardin expression of vascular smooth muscle cells was shown to be regulated by a signaling cascade that involved ROCK, p38 MAPK, PP1a, CPI-17, and myocyte enhancer factor-2 (MEF-2) (Iwasaki et al. 2008).
Consequently, ROCKs may contribute to critical cellular mechanisms in many CVDs. Inhibition of ROCKs prevents smooth muscle proliferation induced by platelet-derived growth factor, thrombin, or urotensin-II that has been identified to involve ROCK-induced upregulation of the cyclin-dependent kinase inhibitor p27Kip1 (Liu et al. 2011).
In the endothelium, ROCKs are important regulators of angiogenic migration and permeability and barrier function. ROCKs have also been shown to exert a negative effect on NO production by inhibition of endothelial NO synthase (Bivalacqua et al. 2004), while endogenous NO can inhibit the RhoA/ROCK pathway (Noma et al. 2012).
ROCK inhibitors, such as the pyridine derivative Y-27632 and fasudil, have been demonstrated to normalize arterial pressure in a rat model of arterial hypertension (Tsounapi et al. 2012) as well as human pulmonary hypertension (Li et al. 2009). Upregulation of ROCKs can be observed in arteriosclerotic lesions (Kandabashi et al. 2000, 2002) and contributes to lesion formation in mice (Mallat et al. 2003). ROCK pathways were also shown to be involved in vascular aneurisms (Wang et al. 2005), myocardial ischemia (Bao et al. 2004), and ventricular remodeling after myocardial infarction (Hattori et al. 2004), and the still-growing list of cardiovascular pathophysiological mechanisms involving ROCKs indicates the central role of this protein kinase family in CVD.
1.2.3 Enzymatic and Nonenzymatic Glycation
Glycosylation is a posttranslational protein modification effected by site-specific enzymatic addition of saccharides. Despite the still unclear position between adverse and beneficial effects, glycosylation of proteins has been demonstrated to be an important regulatory mechanism in the cardiovascular system. Under healthy, physiological conditions, glycosylation is important for cell-cell adhesion, protein stability (protection from degradation), and regulation of enzyme activity (Spiro 2002). Carbohydrates may be attached to the nitrogen of asparagine and arginine (N-linked), to the hydroxyl oxygen of serine, threonine, hydroxylysine, or hydroxyproline (O-linked); to the phosphate of phosphoserine (P-linked), at the carbon of tryptophan (C-linked); or via phosphoethanolamine to the carboxy-terminus of proteins (glycation). Thirteen different monosaccharides and 8 amino acid types have been identified to form bonds, enabling 31 different combinations (Spiro 2002). While N-linked glycosylation is the most widely distributed form, the compound formed by attachment of β-N-acetylglucosamine to serine and threonine residues (O-GlcNAcylation) has been recognized as a key regulator of many critical biological processes including translation and transcription, nuclear transport, cytoskeletal reorganization, and signal transduction (Laczy et al. 2009). The reaction is catalyzed by O-GlcNAc-transferase (OGT), reversed by O-GlcNAc-hydrolase (OGA), and depends on the substrate UDP-GlcNAc that is generated by the hexosamine biosynthesis pathway (Marshall et al. 1991). Serine/threonine residues of several proteins including endothelial nitric oxide synthase (eNOS) (Du et al. 2001), estrogen receptor-β (Cheng and Hart 2001), RNA polymerase II (Kelly et al. 1993), and c-Myc (Kamemura et al. 2002) were shown to be phosphorylation sites as well as targets of O-GlcNAcylation. These posttranslational modifications are not mutually exclusive, as some proteins can be phosphorylated and O-GlyNAcylated concomitantly (Wang et al. 2007; Yang et al. 2006). The linkage between ROCK, AMPK, and SAPK pathways (Lima et al. 2011; Cheung and Hart 2008) and O-GlcNAcylation indicates that this modification reflects cellular stress in addition to functioning as a metabolic sensor (Ngoh et al. 2010).
While a number of cardioprotective effects of O-GlcNAcylation were reported (Fulop et al. 2007; Porter et al. 2012), O-GlcNAcylation has also been demonstrated to be responsible for adverse effects of diabetes on the heart (Marsh et al. 2011) and vasculature (Du et al. 2001; Lima et al. 2010). Hyperglycemia represents a major risk factor for macro- and microvascular diseases associated with diabetes mellitus (Nathan 1996), and O-GlcNacylation was shown to contribute to the effects of diabetes on vessels (Buse 2006). The molecular mechanisms by which O-GlcNAcylation contributes to endothelial vascular dysfunction have been demonstrated to be based on modification of several important regulatory proteins that govern blood vessel function, including PGI2 synthase (Du et al. 2006) and several transcription factors (including specific protein 1, activator protein-1, cAMP response element-binding protein-1, host cell factor-1, nuclear factor of activated T cells, tumor suppressor gene-53) that regulate the expression of proteins like plasminogen activator inhibitor 1 (PAI-1) (Du et al. 2000), thrombospondin-1 (TSP-1) (Raman et al. 2007), and angiopoietin-2 (Ang-2) (Yao et al. 2007). PAI-1 inhibits serine proteases, attenuating the fibrinolytic system in blood and supporting clot formation (Van de Werf et al. 1984). High TSP-1 levels are made responsible for mesangial cell proliferation and increased cellular matrix protein production (Murphy et al. 1999; Yevdokimova et al. 2001), and Ang-2 is an antagonist of Ang-1-promoted Tie2 signaling, leading to an attenuation of blood vessel maturation and stabilization (Chen and Stinnett 2008). The role of PAI-1, TSP-1, and eNOS in diabetes-associated vascular disease is consistently reflected by identification of several polymorphisms in these genes that are linked to vasculopathy (Saely et al. 2008; Santos et al. 2012; Stenina et al. 2004). The O-GlcNAcylation of eNOS at serine 1177, the site for phosphorylation and activation of the enzyme, has been demonstrated to reduce NO production in endothelial cells (Federici et al. 2002). The resulting reduced NO production contributes to impaired relaxation of vessels (Lima et al. 2010). O-GlcNAcylation-mediated decrease in eNOS activity was also shown to be associated with an increase in matrix metalloproteinase activity, an imbalance implicated in diabetes (Galis and Khatri 2002). Matrix metalloproteinases are additionally known to cleave and activate precursors of vasoactive proteins like endothelin 1 (Fernandez-Patron et al. 1999) and cytokines like TNF-α (Gearing et al. 1995) and may thus further impair relaxation and promote inflammatory responses (Fig. 1.2).
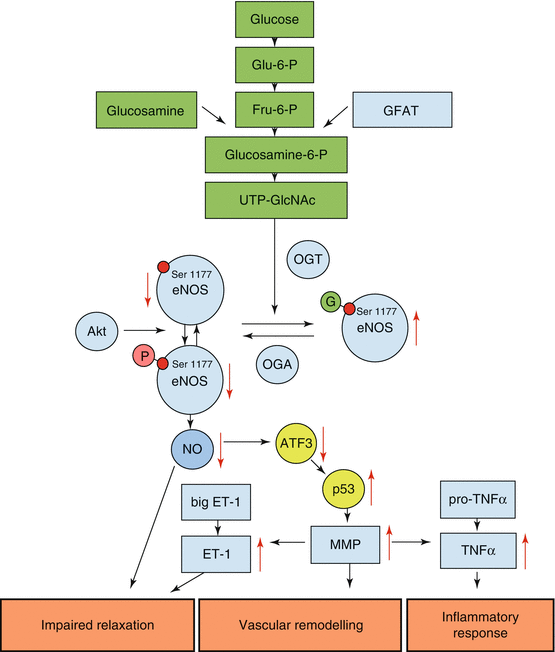
Fig. 1.2
Schematic illustrating molecular mechanisms mediating the adverse effects of O-GlcNAcylation of eNOS on diabetic vasculature. UDP-GlcNAc is produced by the hexosamine biosynthesis pathway (green) and transferred to eNOS at serine 1177, an enzyme phosphorylation and activation site, by b-N-acetylglucosaminyltransferase (OGT). This modification reduces eNOS activity and NO production in endothelial cells and is contributing to impaired vessel relaxation. In addition, a decrease in eNOS activity leads to activation of the cellular stress-induced transcription factor p53 by reduced activity of its inhibitor ATF3 and enhanced expression and activity of matrix metalloproteinases. These metalloproteinases can directly affect vascular remodeling but may also cleave and activate precursors of vasoactive factors and cytokines that contribute to vascular dysfunction. Glu–6–P glucose-6-phosphate, Fru–6–P fructose-6-phosphate, glucosamine-6-P glucosamine-6-phosphate, UDP-GlcNAc UDP-N-acetylglucosamine, OGA β-N-acetylglucosaminidase, Akt Akt kinase, protein kinase B, ATF3 activating transcription factor 3, p53 tumor protein 53, ET–1 endothelin-1, MMP matrix metalloproteinase, TNF–α tumor necrosis factor α
Advanced glycation end products (AGE) are products of the reaction of saccharides with lipids, nucleic acids, and proteins. They are generated by a nonenzymatic process, termed “Maillard reaction.” Over months and years, the initial products undergo various changes until finally yielding a stable AGE compound. Consequently, AGEs accumulate by age and are considered as biomarkers of senescence (Simm et al. 2007).
Epidemiological Examples Demonstrating Relevance of Glycation
AGEs mediate their effects mainly by cross-linking extracellular matrix (Francis-Sedlak et al. 2010; Kamioka et al. 2011) and intracellular proteins (Bidasee et al. 2003, 2004) and also by affecting intracellular glycation and activation of specific receptors (Singh et al. 2001). Binding of AGEs to their cellular surface receptors (RAGE, Neeper et al. 1992), as expressed in endothelial cells, smooth muscle, and monocytes, promotes vasoconstriction, atherogenesis, and inflammation as associated with coronary dysfunction, thrombosis, and atherosclerosis (Hartog et al. 2007). A polymorphism in RAGE has been found associated with the onset of diabetes, atherosclerosis, and renal dysfunction in patients with hypertension (Kawai et al. 2013).
Plasma levels of pentosidine, a cross-linking AGE, have been demonstrated to be a marker of severity and prognosis in heart failure patients (Koyama et al. 2007; Raposeiras-Roubin et al. 2010). In addition to their involvement in coronary artery disease, AGEs were shown to be involved in carotid stenosis and peripheral artery occlusive disease as well as diabetic microangiopathies (Chen et al. 2004; Gerrits et al. 2008; Murata et al. 1997).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


