Figure 2–1.
Schematic cardiac electrical activity, from molecule to patient channel: single channel current versus time recorded using the patch-clamp technique, upward inflection of the signal indicates outward current through a single channel; cell: outward K+ current through the open K+ channels present on the cell membrane; tissue: action potential resulting of the activity of all the different channels of a myocyte, recorded using the voltage-clamp technique; human: EKG resuming the electrical activity of the different regions of the heart
Basic Principles of Cardiac Electrophysiology: From Ion Channels to Ion Currents, Action Potentials and the EKG
Ion Channels
The cell membrane is made of lipids and as such is a perfectly hydrophobic milieu, across which hydrophilic ions cannot directly cross. To penetrate the cell membrane, ions need to find hydrophilic pathways, which are formed by specialized proteins (the ion channels). It happens that the ion channel pathway is not permanently available but transiently flips between open and closed states. Once a hydrophilic pathway is available (the channel is open), ions move passively across the cell membrane depending their respective electrochemical gradient (Fig. 2.2). If the gradient for a given ion species is directed inward, ions enter the cell. If the gradient is outward, ions leave the cell. “Electrochemical” means that two independent forces can move ions across the membrane: the electrical gradient and the chemical gradient. The chemical gradient causes ions to move from a compartment of higher concentration to a compartment of lower concentration (down their chemical gradient; K+ ions move from the intracellular to the extracellular compartment; Na+ ions move from the extracellular to the intracellular compartment). The electrical gradient causes ions to move in a direction opposite to their charge. A negatively charged compartment will attract positively-charged cations but reject negatively-charged anions. In some cases, the electrical gradient and the chemical gradient can oppose each other and eventually be equal; in this situation the force promoting the movement of an ion in one direction equals that promoting its move in the opposite direction. Equilibrium is thus reached. Because the transmembrane potential determines the electrical gradient, the equilibrium potential is the transmembrane potential at which the electrical gradient perfectly opposes the chemical gradient and permits the equilibrium of an ion species. In a cardiac cell, the equilibrium potential is around −98 mV for K+ ions; i.e. at −98 mV (inside negative compared to the outside), the force related to the chemical gradient (directed outward) equals the force related to the electrical gradient (directed inward). The equilibrium (or reverse) potential for each ion species is given by the Nernst equation:
![$${\text{E}}_{\text{i}}=\text{R}\ \text{T}/\text{zF}\mathrm{ln}\left(\text{Extra}[\text{i}]/\text{Intra}[\text{i}]\right)$$](/wp-content/uploads/2016/11/A127647_2_En_2_Chapter_Equ0002a.gif) where Ei is the equilibrium potential for the ion i; R, the thermodynamic gas constant; T, the absolute temperature; z, the charge/valence of the ion i; F, the Faraday (96,485.309 C/mol); Extra [i], the extracellular concentration of the ion i and Intra [i], the intracellular concentration of the ion i.
where Ei is the equilibrium potential for the ion i; R, the thermodynamic gas constant; T, the absolute temperature; z, the charge/valence of the ion i; F, the Faraday (96,485.309 C/mol); Extra [i], the extracellular concentration of the ion i and Intra [i], the intracellular concentration of the ion i.
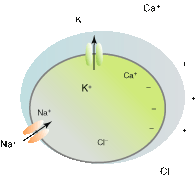
Figure 2–2.
Schematic representation of the chemical and electrical transmembrane gradients of the cardiomyocyte. Font size is proportional to ion concentration
Membrane Resting Potential
A membrane that exclusively conducts a single ion species (i.e. K+ ions), will polarize the transmembrane potential to the equilibrium potential for that ion species (i.e.–98 mV for K+ ions). In the cardiac cell, the sodium-potassium pump enriches the cell with potassium and depletes it for sodium. It also happens that a cardiac cell at rest is predominantly permeable to K+ and thus the resting membrane potential of −80 mV is close to the K+ equilibrium potential. In general, a membrane potential depends on its relative permeability to different ion species. During the action potential (see below), the cardiac membrane potential is no longer exclusively permeable to K+ ions, but becomes permeable to other ion species, causing it to diverge from −80 mV.
Ohm’s Law
One cannot escape Ohm’s law: V = RI where V is the voltage, I the current, and R the resistance of the cell membrane. It means that currents generated by ion movements through ion channels surrounded by an electrical insulator (the cell membrane) affect the membrane potential depending on the membrane resistance. Most ion channels expressed in the heart are voltage-dependent; i.e. they open or close in response to changes in membrane voltage. In most cases, channels open when the cell depolarizes (inside less negative in relation to the outside). Channel opening is often not instantaneous but usually that takes time (as much as hundred of milliseconds in some cases). Thus, most cardiac ion channels are both voltage and time-dependent.
When an ion channel opens, it generates an ion current, which affects the voltage (Ohm’s law). This in turn affects channel opening (voltage-dependence) but not instantaneously (time-dependence). Thus, the electrical activity of the cell should be considered in a tri-dimensional space: voltage, current, and time. If positively charged ions (i.e. cations) enter the cell, this movement creates an inward current that depolarizes the cell membrane. If positively charged ions leave the cell, this movement creates an outward current, which hyperpolarizes or repolarizes the cell membrane if previously depolarized.
Activation/Inactivation
Most cardiac ion channels are governed by two independent processes: activation and inactivation. The cardiac sodium channel is illustrated schematically in Fig. 2.3. At the level of the resting potential (−80 mV), the driving force for sodium ions is clearly in the inward direction (both electrical and chemical gradients are inwardly directed; the equilibrium potential for sodium ions is around +70 mV) but because sodium channel are closed, no current is generated. If the transmembrane potential is brought to values more positive than approximately −60 mV, the threshold potential, sodium channels open (voltage-dependence) and are said to activate, thus generating sodium channel current (INa). Activation is not instantaneous but takes a few tenths of millisecond (time-dependence). Sodium channel activation creates an inward current that further depolarizes the cell. This in turn further recruits sodium channel activation (positive feedback). The positive feedback loop is interrupted when Na+ channels inactivate. Indeed, if the membrane is maintained depolarized, sodium channels do not remain open but close spontaneously. Inactivation is a process that is independent of activation. Thus, in response to membrane depolarization, sodium channels undergo rapid activation and then (less) rapid inactivation. Because activation is faster than inactivation, sodium channels transiently generate an inward (i.e. depolarizing) current. If the membrane is subsequently repolarized to the resting potential, the activation gate closes (this process is called deactivation) whereas the inactivation gate reopens (this process is called reactivation or removal of inactivation): the channel is ready to open in response to a new depolarization stimulus.
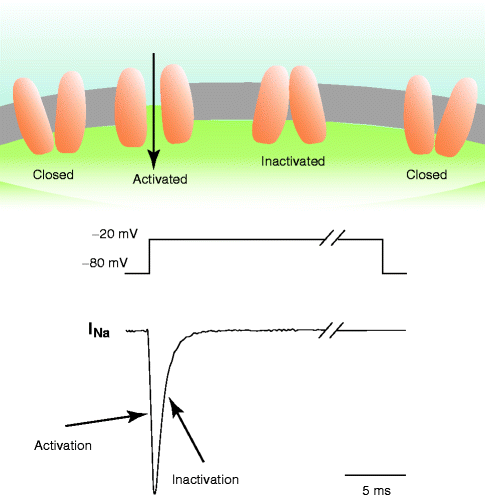
Figure 2–3.
Activation and inactivation of the Na+ current linked to the channel conformation. Schematic Na+ current (bottom) recorded during a voltage pulse (middle). Activation of the current corresponds to an increasing number of Na+ channels in the open conformation (top) due to the depolarization, inactivation corresponds to an increasing number of channels passed from the open to the inactivated conformation. Repolarization allows the inactivated channels to restore the closed conformation
Action Potential
Let’s suppose that for a very few milliseconds cell membrane is much more permeable to sodium than to potassium ions (a large number of Na+ channels activates) (Fig. 2.4). In this situation, the membrane potential is immediately attracted toward the equilibrium potential for Na+ ions (about +70 mV). This occurs during the initial phase of the action potential (phase 0) where the membrane potential is abruptly driven to positive values (inside positive relative to outside). The membrane potential crosses the zero line (this is called an overshoot). Depolarization caused by activation of Na+ channel activates in turn other ion currents such as Ca2+ and K+ currents (see Fig. 2.4), which have slower activation kinetics than Na+ currents. Because the driving force for Ca2+ is inward (between 0 mV and the highly positive equilibrium potential for Ca2+ ions, chemical gradient attracts more Ca2+ into the cell than the electrical gradient limits its entry), activation of Ca2+ channels in response to depolarization generates an inward current, which maintains depolarization and (with other currents) creates the plateau phase of the action potential. Meanwhile, K+ currents are also activated. In this depolarized state, the driving force for K+ ions is outward (above 0 mV, both chemical and electrical gradients attract K+ outside the cell). Activation of K+ channels by membrane depolarization creates an outward current which tentatively repolarizes the cell membrane. During the plateau phase (phase 2), inward currents (mainly Ca2+) equal outward currents (mainly K+); the membrane potential is stable for a few ten milliseconds. The Ca2+ current then progressively inactivates whereas K+ currents progressively activate inducing the cell membrane to repolarize (between 0 and −98 mV, the chemical gradient attracts more K+ outside the cell than the electrical gradient retains it) toward the equilibrium potential of K+ which is about −98 mV. Thus, when Na+ channels are brought to threshold and open, an action potential is generated. The membrane potential is brought positive to −60 mV because an adjacent cell electrically connected through gap junctions undergoes its own action potential process. The electrical signal is thus conducted from cell to cell, from the sinus node down to the ventricular myocardium. The membrane potential can also be brought positive to the threshold potential by an external stimulus (e.g. a pacemaker).
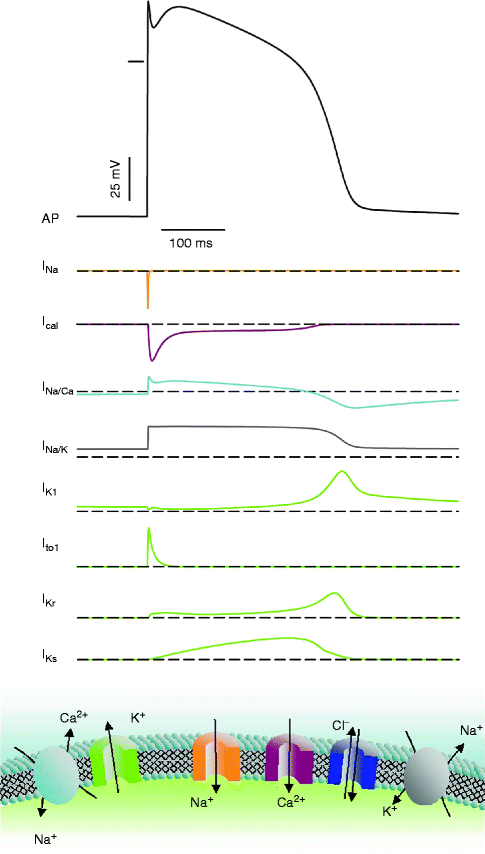
Figure 2–4.
Schematic of a cardiac action potential and underlying ionic currents (top). This is a dynamic process: activation of the Na+ channels generates the voltage upstroke that activates the other ionic currents. The action potential in turn modulates the timecourse of the different currents. Bottom: the different ion channels and transporter species. Arrows denote the direction of the different currents in voltage range of the action potential
Specialized Tissues
Action potentials of cardiac cells, specialized in automaticity (e.g. nodal cells) or in conduction (e.g. Purkinje fibers), have different shapes and characteristics (see Fig. 2.6). Automatic cells from the sinus node or from the atrio-ventricular node have a less polarized membrane potential because they express less background K+ channels. Because of a less negative membrane potential, their Na+ current is permanently inactivated and the depolarizing phase of their action potential relies on the activation of slow Ca2+ currents. As a consequence, the kinetics of the rising phase (a parameter that strongly influences conduction velocity) is much slower than in the myocardium. Another consequence of decreased K+ current is slow depolarization of the cell membrane during diastole, termed phase 4 depolarization, bringing the membrane potential to reach spontaneously the activation voltage for Ca2+ current and thereafter to generate an automatic action potential. Inversely, cardiac cells specialized in conduction have a faster rate of rise of phase 0 (dV/dt max) and faster conduction velocity.
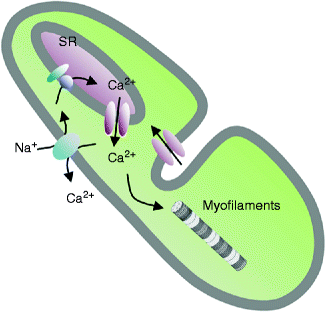
Figure 2–5.
Schematic representation of the cellular mechanisms relating the action potential to contraction. Ca2+ enters the cell when L-type Ca2+channels are activated. The local intracellular Ca2+ concentration ([Ca2+]i) increase activates Ca2+-dependent channels localized on the sarcoplasmic reticulum (SR) membrane (ryanodine receptors), that liberate further Ca2+ into the cytoplasm. A chain reaction induces a general [Ca2+]i increase that induces sarcomeric contraction. At the same time, it induces (i) the Ca2+-dependent inactivation of the sarcolemmal L-type Ca2+ channels (ii) the re-pumping of the intracellular Ca2+ toward the sarcoplasmic reticulum (Ca2+-ATPase) and out of the myocyte (Na-Ca exchanger), resulting in a reduction of [Ca2+]i to baseline diastolic levels
Surface EKG
As already stated in the introduction, the action potential results from the a summation in time of individual channel openings and closings. Multiple action potentials arising from the different segments of the heart are integrated in time and space to generate the extracellular signal known as surface EKG (Fig. 2.6). Because of this continuum, one might expect that an alteration in channel function can cause an anomaly of the action potential shape and therefore a change in the surface EKG. The long QT syndrome is such a situation where a loss-of-function mutation in a K+ channel gene involved in cardiac repolarization prolongs the action potential duration (less net outward current is available to repolarize the cell membrane) and prolongs the QT interval.
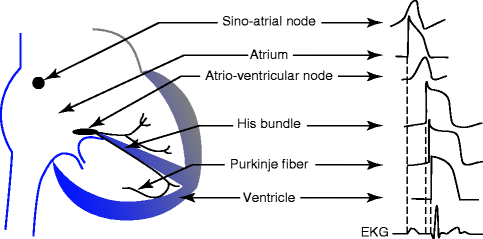
Figure 2–6.
Schematic showing the diversity of action potentials morphologies in different regions of the heart. Dotted lines show correspondence of the activation of the different action potentials to the surface EKG
Excitation-Contraction Coupling
Coupling between the electrical stimulus and contraction is ensured through movements of Ca2+ in and out of intracellular stores [1, 2]. The main store for Ca2+ in a cardiac cell is the sarcoplasmic reticulum where Ca2+ is buffered with specialized proteins such as calsequestrin. Diastolic free Ca2+ in the cytosol is maintained very low in the order of 10−7 M. During the action potential, a small amount of Ca2+ entering the cell through L-type Ca2+ channels, triggers the release of Ca2+ from the sarcoplasmic reticulum through Ca2+-release channels (these are intracellular ion channels labeled by their high affinity for the alkaloid ryanodine) located in the sarcoplasmic reticulum membrane (Fig. 2.5). This mechanism called Ca2+-induced Ca2+-release, produces a relatively large release of Ca2+ from the sarcoplasmic reticulum and invasion of the cytoplasm with free Ca2+. Increased cytoplasmic Ca2+ binds to troponin, opening the myosin binding sites on filamentous actin, and force is produced. Decreased cytoplasmic Ca2+ resulting from its recapture in the sarcoplasmic reticulum by Ca2+-ATPase located in the sarcoplasmic reticulum membrane produces relaxation. Because the system is in perfect equilibrium, the exact amount of Ca2+, which has entered the cell via L-type channels, is now extruded out of the cell through the Na-Ca exchanger.
Gene Correlates for Cardiac Ion Channels
Completion of the sequencing of various genomes including human and mouse has resulted in the elucidation of the complete repertoire of ion channel genes. The human genome contains approximately 25–30,000 genes. Alternative splicing should produce at least 3-times as many mRNA transcript species. However, a specialized cell, e.g. a cardiomyocyte, expresses only 1/3 of the whole genome although this proportion may not be a fixed amount but may vary with development. The human and mouse genomes contain about 230–250 genes encoding ion channel α-subunits and β-subunits. However, as stated above the heart does not express the entire collection of ion channel genes but only a subset. This dramatic progress in our knowledge of mammalian genomes stimulated high-throughput methods (microarrays, sequencing, real-time PCR), which provide information on ion channel expression at a genome scale in various physiological and pathophysiological situations. As example, we addressed regionally ion channel expression in the non-diseased human heart with a genomic approach and highlighted significant differences (Table 2.1), with potentially important implications for understanding regional electrophysiology, arrhythmia mechanisms, and responses to ion channel blocking drugs. Concordance with previous functional studies suggests that regional regulation of cardiac ion-current expression may be primarily transcriptional. However, the processes that govern ion-channel function are complex (Fig. 2.7). Gene transcription controls the production of mRNA, which is then transported out of the nucleus to be translated into proteins. After extensive processing to optimize folding and promote trafficking by specialized chaperone proteins, the mature ion-channel protein is inserted into the cell membrane. Misfolded proteins are targeted for degradation by cellular-control machinery like proteasomes. In the cell membrane, proteins are subjected to functional regulation by enzymes like kinases and proteases, which govern the phosphorylation of key amino acids that control ion-channel activity. The channel protein can also be internalized and targeted for degradation in lysosomes or recycled back to the cell membrane. An additional layer of regulation is provided by micro-RNAs (miRNAs or miRs), which can suppress channel protein-expression by promoting mRNA degradation (which will be reflected in gene-expression profiling as reduced mRNA levels) or by repressing protein translation (which will not). Over the last few years, there has been an extensive miR research showing that miRs could be pivotal regulators in heart normal development and cardiac physiology, as well as in disease development.
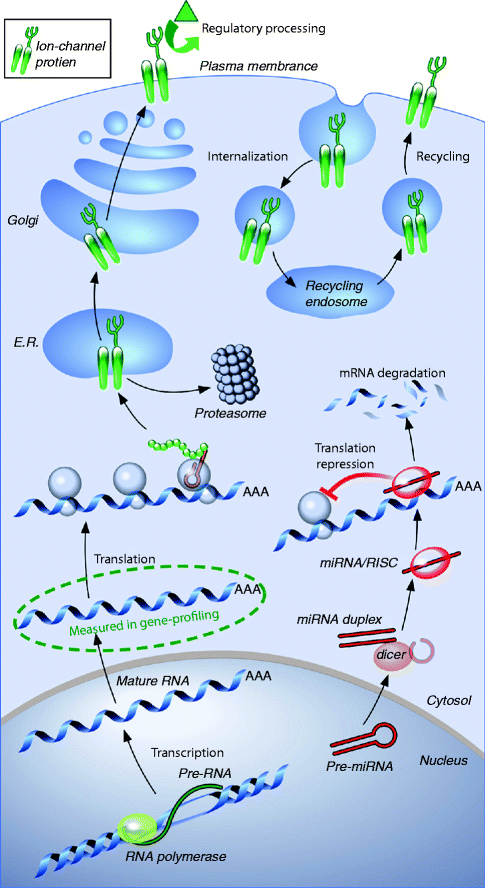
Table 2–1.
Principal ionic currents, channels and auxiliary subunits, and genes expressed in the human heart
Current | Name | α-subunit or transporter | Gene | Auxilary sub-unit | Gene | Remarks |
|---|---|---|---|---|---|---|
INa | Na+ current | Nav1.5 | SCN5A | β1 – β3 – β4 | SCN1B – SCN3B – SCN4B | TTX insensitive channel |
Nav1.7 | SCN9A | Channel in Purkinje fibers only | ||||
Nav1.3 | SCN3A | in atrial cells only | ||||
Ito fast | Fast transient outward K+ current | Kv4.3 | KCND3 | KChiP2 – KChaP – DPP6 | KCNIP2 – PIAS3 – DPP6 | KChiP2 gradient in ventricle |
Ito slow | Slow transient outward K+ current | Kv1.4 | KCNA4 | Kvβ1.2 – Kvβ2 | KCNAB1 – KCNAB2 | |
ICaL | L – type Ca2+ current | Cav1.2 | CACNA1C | β2 – α2δ1 and 2 | CACNB2 – CACNA2D1 and D2 | Channel absent in ventricle |
Cav1.3 | CACNA1D | |||||
ICaT | T-type Ca2+ current | Cav3.1 | CACNA1G | Absent in ventricle | ||
IKr | Fast delayed rectifier K+ current | HERG | KCNH2 | MiRP1? | KCNE2? | |
IKs | Slow delayed rectifier K+ current | KvLQT1 | KCNQ1 | minK (MiRP3) | KCNE1 (KCNE4) | Gradient in ventricle |
IKur | Ultra-rapid outward K+ current | Kv1.5 | KCNA5 | Kvβ1.2 – Kvβ1.3 | KCNAB1 | Channel absent in ventricle |
IK1 | Inward rectifier K+ current | Kir2.1 | KCNJ2 | Mostly in ventricle, absent in node cells | ||
Kir2.2 – Kir2.3 | KCNJ12 – KCNJ4 | Absent in node cells | ||||
IK, Ach | Acetylcholine-dependent K+ current | Kir3.1 – Kir3.4 | KCNJ3 – KCNJ5 | Absent in ventricle | ||
INa/K | Na-K pump current | Na-K pump | ATP1A | Responsible for K+ and Na+ gradient | ||
INa/Ca | Na-Ca exchanger current | Na-Ca exchanger | SLC8A1 | Contributes to Ca2+ gradient | ||
If | Pacemaker current | HCN2 – HCN4 | In nodal tissue |
Figure 2–7.
Molecular factors determining ion-channel function. Transcription of ion-channel subunit messenger-RNA (mRNA) is followed by export from the nucleus and translation into protein. The protein then undergoes a series of maturation steps in the endoplasmic reticulum (E.R.) and Golgi apparatus before final trafficking to the cell membrane. At the membrane, regulatory changes (e.g. control of phosphorylation) can alter channel function, and the protein can be internalized and either destroyed or recycled to the surface. Subunit-expression can also be altered by micro-RNAs (miRNAs, or miRs), which can promote mRNA degradation or impede translation by directing RNA-silencing complexes (RISCs) to target mRNA (From Nattel et al. [3]. Reprinted with kind permission from Elsevier)
Technologies to Explore Cardiac Electrophysiology
Although the first recording of a cardiac action potential was obtained about 50 years ago [4], it is the discovery and development of the patch-clamp technique (which brought the Nobel prize of medicine to E. Neher and B. Sakman in 1991), which has been key to our understanding of cardiac cellular electrophysiology [5, 6]. The technique is applied to isolated cells either freshly dissociated from cardiac tissues or maintained in culture. Depending the configuration used, the patch-clamp technique can record the electrical activity of a single channel molecule (amplitudes of a single channel current are in the order of a picoA = 10−12 A) or the electrical activity of the ensemble of channel protein expressed in a whole cell (current amplitudes may be in the order of one nanoA = 10−9 A). At the level of a single protein recording, the channel appears in a binary situation either close or open. Voltage-dependent activation is visible because the channel spends more time in the open configuration and thus less in the close configuration in response to a voltage step (usually depolarizing). Inactivation is also visible as a progressive decrease in channel firing with time at a stable voltage (see Fig. 2.3). In the current-clamp mode, variations in the voltage (action potentials) are measured whereas in the voltage-clamp mode, ion currents are measured.
Another major technical step in cardiac electrophysiology has been inherited from molecular biology techniques and relates to our capacity to make host cells (e.g. COS-7 or HEK cells) maintained in culture to express foreign genes. Cells are transfected with ion channel cDNA using routine non-viral methods and then patch-clamped after 24–72 h in culture. Recombinant ion channel proteins (in particular of human origin) are available for physiological and pharmacological investigations [7]. Site directed mutagenesis permits investigations of mutated constructs shading light into genotype-phenotype relations [8].
Genetic manipulation of ion channel genes and regulators have been successfully achieved in the mouse with either transient or permanent over-expression or invalidation of target channels [9, 10] and provided appropriate models to study complex channelopathies mechanisms [11], with important limits, though [12]. Development of in vivo electrophysiological methods adapted to the very small size of this animal (a mouse heart is about 100 mg) has provided similar investigations capacities as in human [13–15].
Expression of recombinant foreign channel proteins in host cell systems, as well as genetic invalidation of ion channel genes in the mouse, have been instrumental to correlate cardiac ion channels (and ion currents) with their encoding genes. In addition, these investigations have also shown that ion channels do not express in isolation in the cell membrane but rather in concert with other regulatory proteins within a channel complex. Identification of missing members of ion channel complex is the subject of active research in various laboratories with the objective to target novel candidate genes for cardiac channelopathies.
Cardiac Cellular Electrophysiology of the Human Heart
The patch-clamp technique applied to single cells dissociated from cardiac biopsies sampled during open-heart surgery in association with molecular biology techniques has provided an impressive body of information on the cellular electrophysiology of the human heart.
At the atrial level, the human action potential is initiated by a fast-activating fast-inactivating Na+ current carried by Nav1.5 channels (encoded by the SCN5A gene) in association with its β1 subunits (SCN1B) [16, 17]. Other Na+ channel α-subunits including Nav1.3 (SCN3A) [18], Nav2.1 (SCN6A) [19] and β3 and 4-subunits (SCN3B and SCN4B, respectively) [20] are also expressed in the human atrium although at a much lower level than Nav1.5 which is by far the predominant cardiac Na+ channel. The Nav1.5 carried Na+ current is responsible for the upstroke of the action potential (phase 0) and carries energy for fast conduction. Fast depolarization triggers the activation of transient outward and inward currents. The transient outward current produces initial repolarization of the action potential and a clearly visible notch inscribed prior to the AP plateau. Transient outward current is made predominantly by Kv4.3 channel (KCND3) [21] in association with its regulatory β-subunits KChiP2 [22] and, putatively, DPP6 [23, 24]. Because inactivation of the transient outward current is fast, this current determines the level of the plateau phase and therefore influences the activation of other currents but does not directly influence phase 3 repolarization kinetics. Transient Ca2+ currents provide inward current to maintain the cell depolarized during the plateau. Two types of Ca2+ currents are operative: L-type (long lasting), which are target for calcium channel blockers, and T-type (fast inactivated). L-type Ca2+ currents are predominantly carried by Cav1.2 (CACNA1C) [25] and to a much lower extent by Cav1.3 (CACNA1D) [26] channels in conjunction with their auxiliary subunits: Cavβ2 [25], Cavα2δ2 [27], Cavα2δ1 [28]. T-type Ca2+ currents are brought by Cav3.1 channels (CACNA1G) [29]. Repolarization of the action potential (phase 3) is initiated by the delayed rectifier K+ current, which has two components: a fast activating component termed IKr which is carried by HERG (KCNH2) channels and MiRP1 (KCNE2), its β-subunits [30] and a slow component IKs which is carried by KvLQT1 (KCNQ1) channels in association with the regulatory β-subunits minK (KCNE1), MiRP1 and MiRP2 (KCNE3) [31] or the A-kinase anchor protein 9 (AKAP-9) [32]. An ultra-rapid K+ current (activation is 2 fold faster than IKr) is specific of the atrium in human and is carried by Kv1.5 channels (KCNA5) [33, 34]. Final repolarization is achieved by background time-independent currents (also called inward rectifiers), which are also responsible to maintain a negative membrane polarization during diastole. Kir2.1 channels (KCNJ2) are less abundant than in the ventricle accounting for the less negative resting potential in the atrium [35]. Atrial myocytes also express Kir2.2 and Kir2.3 channels (KCNJ12 and KCNJ4). Specific of the atrium are Kir3.1 and Kir3.4 channels (KCNJ3 and KCNJ5), which open in response to cholinergic stimulation and shorten the action potential duration [30]. Other background K+ currents include TWIK1 (KCNK1) and TASK1 (KCNK3) currents [36]. The cardiac function of another class of ion channel is only now beginning to emerge and concerns the transient receptor potential (TRP) channels, especially the TRPC subclass. TRP channels permeate many different cations, and are most likely critical regulators of microdomain Ca2+ signaling in the heart in disease states such as hypertension, cardiac conduction block and cardiac hypertrophy [37]. TRPC1 and TRPC6 channels are key players in muscle mechanotransduction [38]. Other currents are related to the Na-K pump (ATP1A1) [39], which generates an outward current and the Na-Ca exchanger (SLC8A1), which generates an inward current and participates to maintain the plateau. The role of chloride channels in the human heart is still ascertained. Electrical connection between cells is ensured by the expression of connexin channel proteins (connexin 40 (GJA5) is the main isoform in atria while Cx43 (GJA1) characterizes the ventricles) [40].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


