Fig. 6.1
A scheme of short- and long-term changes in NGF expression and nerve density in animal models of myocardial injury (Reproduced with permission from Govoni et al. [16])
6.6 From Acute Myocardial Injury to Chronic Heart Failure through Cardiac Remodeling
As already reported before, experimental evidence demonstrated a progressive decrease of NGF plasma level in rats 8 weeks after a coronary occlusion, following a prolonged catecholamine effect in an advanced heart failure [23]. Similarly, prolonged infusion of noradrenaline in rats is associated to a reduction of NGF and of its receptor TrkA, following a decrease in mRNA transcription [24] (Fig. 6.2). It seems that sympathetic cardiac innervation is firmly regulated by NGF expression and prolonged exposure to high catecholamines, as it happen in advanced heart failure (HF), may induce a reduction of NGF and a consequent decrease in sympathetic fibers density. Several studies also demonstrated that high catecholamine levels reduce metaiodobenzylguanidine (MIBG) uptake in HF, according to two possible mechanisms [25]. The former one implies a competitive reaction between high noradrenaline and MIGB reuptake. The latter one, much more reliable, implies a real cardiac denervation as a cause of a reduced MIGB reuptake. It is reasonable to hypothesize that sympathetic functional denervation comes along with anatomic denervation, which can be seen only in an advanced HF stage after a prolonged catecholamine expression. The negative prognostic value of a depressed MIBG reuptake in patients with HF has been clearly documented by the ADMIRE HF study [26]. In this study indeed a depressed MIBG reuptake identified patients at higher arrhythmic risk and/or at higher risk of an unfavorable progression of the disease.
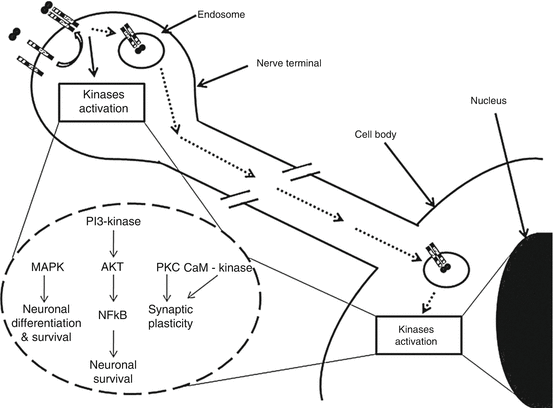
Fig. 6.2
Molecular events following NGF interaction with TrkA receptor in nerve terminals, possibly leading to neuronal remodeling (Reproduced with permission from Govoni et al. [16])
Recent studies stressed the evidence of NGF expression on cardiac myocytes. Meloni et al. [16] observed NGF and TrkA expression on hearts affected by a recent fatal myocardial infarction: NGF was found in cardiomyocytes, and both cardiomyocytes and endothelial cell express TrkA. In the same experimental research, antibodies anti-NGF used in infarcted rats’ hearts cause a lower angiogenesis, an increased apoptosis, and a worsened cardiac function. On the contrary, genetic NGF stimulation improved endothelial cell survival rate and promoted angiogenesis, ameliorating cardiac function 14 days after a heart attack.
In the complex system of neurohormonal interactions discussed above, NGF seems to have a protective role on the ischemic and failing heart, causing maybe also a paradox dangerous effect by increasing too much sympathetic innervation. The key factor to understand this process might involve heterogeneous NGF expression leading to adjacent areas of hyper and hyposympathetic innervation. This anatomical heterogeneity would result and in a marked increase in electrical inhomogeneity at the time of a sudden rise in sympathetic discharge to the heart.
To this last regard, we can speculate that molecules able to positively modulate NGF expression, as already described for other genes, may represent a novel pharmacological strategy that can contribute to counteract some of the negative effects associated with NGF decline and potentially favor cardiac repair after an acute ischemic insult. Conversely, molecules able to reduce NGF expression might be useful to prevent the nerve-sprouting phenomenon, occurring just after MI, which seems involved in the genesis of arrhythmias [27].
6.7 Molecular Pathophysiology of Cardiac Sympathetic Innervation
The extent of myocardial innervation by autonomic neurons is often overlooked when discussing the role of the adrenergic system in cardiac biology or pathology. As discussed above, both the atria and ventricles are densely innervated by sympathetic and parasympathetic autonomic neurons; in the normal heart, sympathetic neurons spread throughout the different regions of the heart with a well-defined distribution pattern. Myocardial innervation depends on a number of factors released by cardiomyocytes, cardiac fibroblasts, and endothelial cells that include neurotrophins such as NGF, neurotrophin-3, and endothelin-1 (ET-1) [28–30]. In addition to such molecules sustaining neuron growth and viability, chemorepulsive factors of the semaphorin family, like Sema3, are secreted by cardiac cells and participate to the guidance of sympathetic axons throughout the myocardium [31]. In the mouse heart, a transmural distribution pattern is established with higher neuronal density in the subepicardial than in the subendocardial areas, and in Sema3a-knockout mice, the failure to develop the correct innervation pattern increased arrhythmic propensity [32]. The balance between neuron-attractant and neuron-repellent factors released by myocardial cells finely shapes the density of sympathetic innervation throughout the cardiac walls. As described above, remodeling of cardiac sympathetic innervation is determinant in the pathogenesis of heart failure following ischemic damage. The neurotrophic factor GDNF (glial cell line-derived neurotrophic factor) was able to increase cardiac innervation in vivo in rats after cardiac cryoinjury and counteracted the progress to HF [33]. This extends the observations of earlier work, in which direct NGF injection into the stellate ganglia neurons was able to increase the function of sympathetic innervation, NE reuptake, and improve cardiac function in an animal model of HF [34], and conversely, genetic knockdown of the NGF gene leads to worsened prognosis after myocardial infarction, due to reduced sympathetic innervation and angiogenesis [35].
The fine architecture of the cardiac neuronal network was recently described using 3-D reconstruction of the distribution of GFP+ cardiac neurons in the intact heart of DBH-GFP mice [36]. When observed at high resolution with methods highlighting adrenergic neurons, axonal “varicosities” can be observed with regular distribution along the neuronal processes (Mongillo M 2015, unpublished). These structures, which represent neuronal sites enriched in secretory vesicles containing noradrenaline and other sympathetic neurotransmitters [37], are found close to the cardiomyocyte sarcolemma, with variable frequency in the different heart regions (Mongillo M 2015, unpublished). Evidence from in vitro models based on sympathetic neurons and cardiomyocyte cocultures demonstrates that direct interaction of the two cell types occurs at specific contact sites and is accompanied by specialization in the cardiomyocyte “postsynaptic” membrane in which β-adrenergic receptors (ARs), together with adaptor/scaffold proteins and intercellular contact proteins, are all specifically enriched [38]. Although with much less definition from the morphological and biochemical standpoint, the interaction site between sympathetic neurons and cardiomyocytes shares features of the well-known neuromuscular junction [39]. The hypothesis of cardiac synapses has been proposed by many authors in the last four decades. It has been suggested that such specific junctions would increase the efficiency of activation of cardiomyocyte b-AR by neuronally released noradrenaline, a requisite for the rapid activation of the cardiac inotropic and chronotropic effects of the fight-or-flight response [40].
Although the details on molecular signaling by beta-adrenergic receptors have been obtained using isolated or cultured cardiomyocytes, it is worth considering them in the context of the cell–cell interaction between sympathetic neurons and cardiomyocytes.
The adrenergic receptor system mediates cardiac responses to the release of catecholamines by the sympathetic neurons. Cardiomyocyte β-1 and β-2 adrenergic receptors (βARs) couple to the activation of cyclic adenosine monophosphate (cAMP) that, mainly through activation of protein kinase A and EPAC [41], operate the increase in rate and force of contraction of the heart. Studies performed in isolated cardiac myocytes showed that the subtype-specific stimulation of the two b-AR isoforms activates a distinct pattern of intracellular downstream targets, with different effects on cardiomyocyte function [42]. While β-1 selective activation exerts potent inotropic and lusitropic effects, β-2 AR have only a minor effect on contractility and fail to enhance relaxation [43]. Furthermore, chronic β-1 AR stimulation caused cardiomyocyte apoptosis [44], and its overexpression in transgenic mice leads to progressive hypertrophy and heart failure [45], while β2AR stimulation showed anti-apoptotic effect [46]. Although both b1 and b2 AR lead to cAMP synthesis, their singular effect has mostly been attributed to partitioning of the receptors on the cell membrane and to the restriction of cAMP diffusion in distinct intracellular compartments [47, 48], where the target effector proteins (e.g., ion channels, SERCa pumps, ryanodine receptors) are localized [49, 15].
Interestingly, direct intercellular contact might also be involved in retrograde cardiomyocyte-to-neuron signaling mediated by neurotrophins. Reduced efficiency of neurocardiac communication, due to alterations in the specific contact structures, would therefore result in decreased activation of b-AR, noradrenaline spillover in the myocardial interstitium, and depletion of noradrenaline stores, all of which are features commonly appreciated in failing hearts [50]. In addition, it would decrease the trophic input to cardiac neurons, which may result, with time, in denervation, also commonly observed in failing hearts [51].
In addition to the role in the acute control over heart rate and contractility, cardiac sympathetic neurons have a role in the regulation of fundamental homeostatic mechanisms in cardiomyocytes. The expression level of the major cardiac ubiquitin ligases, namely, MuRF1 and Atrogin-1 [52], key mediators of the cardiomyocyte ubiquitin–proteasome system, is negatively controlled by b2-AR activation, through PI3K/Akt modulation of the FOXO family of transcription factors [53, 54]. Consistently, selective stimulation of b2-AR causes cardiac hypertrophy, while its inhibition by either receptor antagonists or removal of neuronal input with cardiac sympathetic denervation leads to atrophic remodeling in rodent and rabbit hearts [55]. Interestingly, UPS targets directly regulated by adrenergic receptors including signaling elements involved in the degradation of cAMP, like phosphodiesterases [56] as well as cardiac proteins determinant for cellular coupling and electrophysiology, including Connexin43 [57].
In addition to the pharmacological inhibition of beta-AR, direct modulation of cardiac autonomic nerves has also received considerable attention. Experimental therapies aimed at the shift in autonomic balance toward reduced sympathetic drive and increased vagal activity have been obtained in several ways including pharmacological approaches [58, 59] and direct electrical stimulation. In rats, electrical stimulation of the ear endings and the brainstem exerts an antiarrhythmic effect. Similar results were obtained with electrical stimulation of carotid baroreceptors, which also leads to sympathetic inhibition; chronic vagal stimulation, as elicited by a surgically implantable stimulator [60], has been shown to produce a significant drop in systemic pressure and is currently in clinical trials in drug-resistant hypertension [61]. Given the abundance of data demonstrating the arrhythmogenic potential of sympathetic activity, it seems obvious that the prevention of life-threatening events in patients at high risk of developing VT/VF may represent another clinical application of cardiac sympathetic nerve modulation. To date, however, experimental studies have not adequately addressed this issue.
6.8 Conclusions
The autonomic nervous system responds within seconds to any even minimal cardiac insult. The level of autonomic response might be, in several instances, exaggerated with detrimental consequences of cardiac negative remodeling during and after acute myocardial ischemia. The key issue here is that autonomic responses are the cause and not the consequence of the negative cardiac remodeling (Fig. 6.3). The understanding of the autonomic pathophysiology had eventually critical clinical consequences when, after 25 years of denial toward the use of them, beta-blockers became first-line therapy in heart failure. This understanding needs to be fully appreciated at time like today when the available therapy seems to be unable to further meet the growing challenge of heart failure and direct nerve modulation represents the new hope for such a challenge.
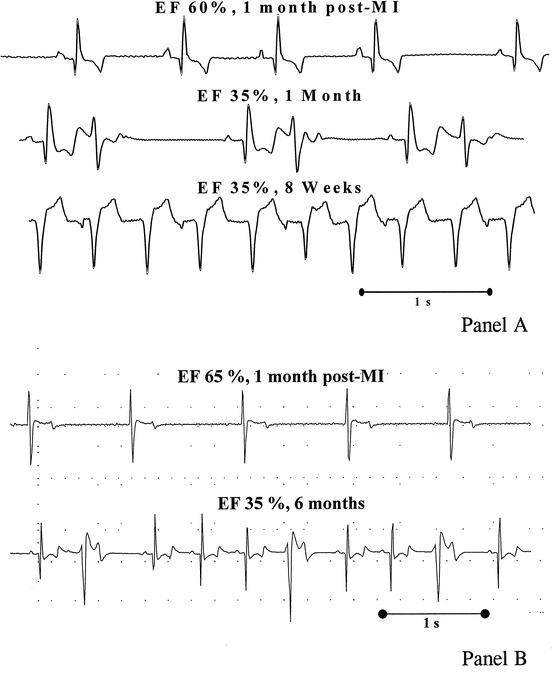
Fig. 6.3
Spontaneous arrhythmias in conscious dogs with a healed anterior MI and circumflex ischemia-dependent progression of left ventricular dysfunction at high and low risk for sudden death. In panel (a) the rapid occurrence of a fatal ventricular tachyarrhythmia in dogs with a depressed vagal control of heart rate is shown. In panel (b) the arrhythmia pattern in a dog with analogous cardiac damage but with preserved vagal reflexes is shown. The level of reflex vagal control determines the fatal or nonfatal consequence of a chronic ischemic LV dysfunction (Reproduced with permission from Adamson and Vanoli [13])
References
1.
2.
Fukuda K, Kanazawa H, Aizawa Y, Ardell JF, Shivkumar K. Cardiac innervation and sudden cardiac death. Circ Res. 2015;116:2005–19.
3.
Berg T, Jensen J. Simultaneous parasympathetic and sympathetic activation reveals altered autonomic control of heart rate, vascular tension, and epinephrine release in anesthetized hypertensive rats. Front Neurol. 2011;2:71.PubMedPubMedCentralCrossRef
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


