Fig. 25.1
Development of human coronary atherosclerosis. The two nonprogressive lesions are intimal thickening or intimal xanthoma (foam cell collections known as fatty streaks, AHA type II). Pathological intimal thickening (AHA type III, transitional lesions) is the first of progressive plaques marked by an acellular lipid pool rich in proteoglycan; inflammation when present is typically confined to the most luminal aspect of this plaque. Fibroatheromas are lesions with areas of necrosis characterized by cellular debris and cholesterol monohydrate with varying degrees of calcification or hemorrhage. Finally, thin-cap fibroatheroma or vulnerable plaques are recognized by their relatively large necrotic cores and thin fibrous caps (Reproduced with permission from reference [1]). LP lipid pool, NC necrotic core, FC fibrous cap
25.2 The Classification of Atherosclerosis
The initial concepts of the progression of atherosclerotic changes were established in the early 1980s where Velican focused on the morphological descriptions of fatty streaks to fibroatheroma and advanced plaques complicated by hemorrhage, calcification, ulceration, and thrombosis [4, 5]. Michael J. Davies, a renowned cardiac pathologist from the United Kingdom who devoted himself to the study of plaque rupture and its associated features, described the characteristics of plaque disruption and the role of inflammation in the development of plaque instability in detail [6, 7]. Similarly Wissler, Strong, McGill, Stary, and many others have contributed to the understanding of atherosclerosis and its relation to risk factors and plaque progression [8–11].
The work of various pioneers, however, failed to note that lesions proceeding rupture share important similarities and yet have not ruptured, i.e., vulnerable plaques or thin-cap fibroatheromas, and we need to direct our focus on detection of this critical phase of atherosclerotic plaque to advance clinical care and facilitate prevention of sudden coronary death. Similarly we need to understand how plaques progress and therefore determine at which phase prevention of further progression will yield the most benefit for reducing CAD. This gap was fulfilled by Stary et al. [12, 13] in the mid-1990s under the auspices of the American Heart Association. These scientists published the first consensus report on the various types of plaques and progression of atherosclerosis. In this consensus classification, lesions were classified into six different numerical categories: early lesions of initial type I, intimal thickening; type II, fatty streak; type III, transitional or intermediate lesion; type IV, advanced atheroma with well-defined region of necrotic core in the intima; type V, fibroatheroma or atheroma with a layer of fibrous connective tissue; and type VI, complicated plaques with surface defects and/or hematoma-hemorrhage and/or thrombosis. At this time it was not appreciated that plaque rupture was not the only mechanism of coronary thrombosis and that plaque erosion occurred in at least 25–30 % of cases and that rarely calcified nodule also caused thrombosis.
Atherogenesis refers to the development of atheromatous plaques within the inner lining (intima) of the arterial wall. Atherogenic dyslipidemia is characterized mainly by three lipoprotein abnormalities: elevated very-low-density lipoproteins (VLDL) and small LDL particles and low high-density lipoprotein (HDL) cholesterol [14, 15]. Many patients with atherogenic dyslipidemia also have an elevated serum apolipoprotein B (ApoB) [16]. Atherogenic ApoB-containing lipoproteins are synthesized from intestinal cholesterol absorption and hepatic cholesterol synthesis (Fig. 25.2). Cholesterol is absorbed from the intestine through ApoB48-containing chylomicrons, while ApoB100-containing very-low-density lipoproteins (VLDL) are produced by the liver. Subsequently, atherogenic ApoB-containing chylomicron remnants, VLDL remnants, and LDL infiltrate into the arterial wall and are taken up by macrophages and smooth muscle cells (SMCs) leading to the generation of atherosclerotic lesions.
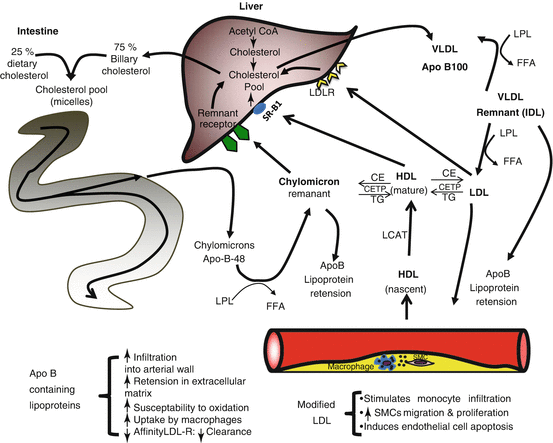
Fig. 25.2
Pathophysiology of atherogenic lipoproteins. Atherogenic ApoB-containing lipoproteins are synthesized from intestinal cholesterol absorption and hepatic cholesterol synthesis. Cholesterol is absorbed from the intestine through ApoB48-containing chylomicrons, which form chylomicron remnants following the hydrolysis of triglycerides (TG) by lipoprotein lipase (LPL) to form free fatty acids (FFA). ApoB100-containing very-low-density lipoproteins (VLDL) are produced by the liver and acted on by LPL to generate VLDL remnants (intermediate-density lipoproteins IDL) and low-density lipoproteins (LDL). Atherogenic ApoB-containing chylomicron remnants, VLDL remnants, and LDL infiltrate into the arterial wall are retained by binding to the extracellular matrix, where the lipoproteins are modified and taken up by macrophages and smooth muscle cells (SMCs) leading to the generation of atherosclerotic lesions. High-density lipoproteins (HDL) can efflux free cholesterol from the atherosclerotic lesions, where cholesterol is esterified by lecithin cholesterol acyltransferase (LCAT) to cholesterol esters (CE), and CE exchanged to ApoB-containing lipoproteins by cholesterol ester transfer protein (CETP). The liver can take up cholesterol from HDL through scavenger receptor B1 (SR-B1), from LDL by LDL receptors (LDL-R), and from chylomicron remnants by remnant receptors. Hepatic cholesterol is excreted into the intestine in bile where it can be reabsorbed into chylomicrons or is returned to circulation in VLDL particles to generate additional atherogenic ApoB-containing lipoproteins
Our laboratory, having the largest registry of human autopsy cases of patients dying from sudden coronary death in the world, reported that the numeric nomenclature was incomplete. Also, at this point it was not appreciated that the precursor lesion of plaque rupture [1] is a thin-cap fibroatheroma (vulnerable plaque) and that other causes of thrombosis existed besides rupture. Furthermore, the atherosclerosis process is considered too complicated to be categorized into numerical groups. These limitations prompted us to develop a modified version of the AHA classification. In our modified classification [1] (Figs. 25.1 and 25.3, Table 25.1), numeric AHA lesion types I to IV were abandoned and replaced by descriptive terminology to include adaptive intimal thickening, intimal xanthoma (fatty streak), pathological intimal thickening, and fibroatheroma, the latter we have further divided recently into early and late fibroatheromas. Lesions referred to as AHA types V and VI were discarded since they failed to account for the three different morphologies of thrombosis (rupture, erosion, and calcified nodule) or their relationship to stabilized plaque that is representative of lesions that have healed and that with time calcify.
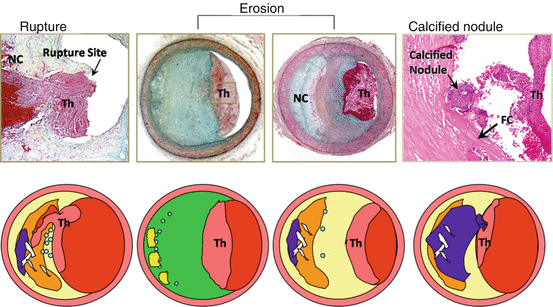
Fig. 25.3
Atherosclerotic lesions with luminal thrombi. Coronary plaque features responsible for acute thrombosis comprise three different morphologies: rupture, erosion, and calcified nodules. Ruptured plaques are thin fibrous cap atheromas with luminal thrombi (Th). These lesions usually have an extensive necrotic core (NC) containing large numbers of cholesterol crystals and a thin fibrous cap (<65 μm) infiltrated by foamy macrophages and T-lymphocytes. The fibrous cap is thinnest at the site of rupture and consists of a few collagen bundles and rare smooth muscle cells. The luminal thrombus is in communication with the lipid-rich necrotic core. Erosions occur over lesions rich in smooth muscle cells and proteoglycans. Luminal thrombi overlie areas lacking surface endothelium. The deep intima of the eroded plaque often shows extracellular lipid pools, but necrotic cores are uncommon; when present, the necrotic core does not communicate with the luminal thrombus. Inflammatory infiltrate is usually absent but, if present, is sparse and consists of macrophages and lymphocytes. Calcified nodules are plaques with luminal thrombi showing calcific nodules protruding into the lumen through a disrupted thin fibrous cap. There is absence of an endothelium at the site of the thrombus, and inflammatory cells (macrophages and T-lymphocytes) are absent (Reproduced with permission from reference [1])
Table 25.1
Modified AHA consensus classification based on morphological description
Description | Thrombosis | |
|---|---|---|
Non-atherosclerotic intimal lesions | ||
Intimal thickening | Normal accumulation of smooth muscle cells (SMCs) in the intima in the absence of lipid or macrophage foam cells | Absent |
Intimal xanthoma | Superficial accumulation of foam cells without a necrotic core or fibrous cap. Based on animal and human data, such lesions usually regress | Absent |
Progressive atherosclerotic lesions | ||
Pathological intimal thickening | SMC-rich plaque with proteoglycan matrix and focal accumulation of extracellular lipid | Absent |
Fibrous cap atheroma | Early necrosis: focal macrophage infiltration into areas of lipid pools with an overlying fibrous cap | Absent |
Late necrosis: loss of matrix and extensive cellular debris with an overlying fibrous cap | ||
Thin-cap fibroatheroma | A thin fibrous cap (<65 μm) infiltrated by macrophages and lymphocytes with rare or absence of SMCs and relatively large underlying necrotic core. Intraplaque hemorrhage/fibrin may be present | Absent |
Lesions with acute thrombi | ||
Plaque rupture | Fibroatheroma with cap disruption; the luminal thrombus communicates with the underlying necrotic core | Occlusive or nonocclusive |
Plaque erosion | Plaque composition, as above; no communication of the thrombus with necrotic core. Can occur on a plaque substrate of pathological intimal thickening or fibroatheroma | Usually nonocclusive |
Calcified nodule | Eruptive (shedding) of calcified nodule with an underlying fibrocalcific plaque with minimal or absence of necrosis | Usually nonocclusive |
Lesions with healed thrombi | ||
Fibrotic (without calcification) | Collagen-rich plaque with significant luminal stenosis. Lesions may contain large areas of calcification with few inflammatory cells and absence of necrosis. These lesions may represent healed erosions or ruptures | Absent |
Fibrocalcific (±necrotic core) | ||
25.3 Intimal Thickening and Fatty Streaks
Intimal thickening (AHA type I) is the earliest change in the arterial wall, which consists of smooth muscle cells in an extracellular matrix. Smooth muscle cell (SMC) proliferation has been observed in the media before birth but is rare after birth, whereas the intimal replication index was only 2–5 % [17]. Although intimal thickening is more frequently recognized in the atherosclerosis-prone arteries (coronary, carotid, abdominal and descending aorta, and iliac artery) [18], it is currently regarded as an adaptive change because the number of proliferating cells is 10- to 20-fold higher in fibroatheromas (AHA type IV) as compared to lesions that show diffuse intimal thickening [19, 20].
The term “fatty streak” or “intimal xanthoma” (AHA type II, fatty streak) is primarily composed of infiltrating lipid-laden cells of either macrophage or SMC origin. Although this lesion is described as the earliest lesion of atherosclerosis in accordance to the AHA classification [21, 22], it does not always progress to the advanced lesion of fibroatheroma and in fact has been shown to regress especially in the thoracic aorta of young individuals [9]. Adaptive intimal thickening lesions can also be found in adults, particularly in those with little atherosclerosis, or in locations of arteries that are resistant to the development of atherosclerosis and, in fact, do not always progress.
25.4 Pathological Intimal Thickening
Pathological intimal thickening (PIT, AHA type III lesion) is defined by us and others as the earliest lesion of progressive atherosclerosis. PIT lesions consist of SMCs within a proteoglycan and collagen-rich (type III) extracellular matrix along with the presence of lipid pools in the vicinity of the media [1]. Within the area of the lipid pool, there is an absence of SMCs, and the matrix is composed of hyaluronan and proteoglycans such as versican along with lipid deposition. In the precursor lesion of PIT, i.e., adaptive intimal thickening, there is presence of proteoglycan biglycan and decorin surrounding the SMCs. As the intima thickens, the SMCs undergo apoptosis close to the media with increasing deposition of basement membrane (BM) surrounding the individual cells. The BM can be visualized by periodic acid Schiff (PAS) staining. Also, apoptotic smooth muscle cell nuclei have been identified by in situ nick end labeling of human coronary and carotid plaques [23]. It has been shown that structural changes in the glycosaminoglycan chain of proteoglycans, i.e., versican and biglycan, are the initial proatherogenic proteoglycans that lead to the binding of atherogenic lipoproteins [24, 25]. In contrast, the internal thoracic artery that is resistant to the development of atherosclerosis has a thin neointima that is rich in decorin [26].
Over time, these PIT lesions show the accumulation of macrophages toward the lumen remote from the lipid pool, although lesions of PIT do not all show the presence of luminal macrophages; PIT lesions with macrophages are considered by us as a more advanced stage of atherosclerosis. Nakashima et al. also showed early coronary lesion progression near branch points [24] consisting of extracellular lipid accumulation underneath a layer of foam cell macrophages. The reasons why macrophages accumulate in PIT lesions, although not fully understood, may be linked to specific proteins being expressed within the lipid pools, which as of today remains unknown; however, apoptotic signals may be involved. We have observed the presence of free cholesterol seen as empty fine crystalline structures in paraffin-embedded sections within the area of the lipid pool to varying degrees, but are never excessive nor long. The precise origin of free cholesterol in PIT lesions remains unknown, but they are likely derived from membranes of dying SMC [27]. It is also conceivable that extracellular lipid accumulation within lipid pools may have originated from the circulating plasma lipoprotein, which have been demonstrated in studies addressing the association of proteoglycans and plasma lipoprotein [28, 29]. Also, experimental studies have demonstrated that intrinsic phenotypic changes in smooth muscle cell at the initial stage of plaque formation in the setting of hypercholesterolemia exert a greater control over SMC apoptosis than cell-cell interactions or the microenvironment of the plaque [30, 31]. In the absence of hyperlipidemia, SMCs are efficient phagocytes of apoptotic SMCs in vitro and in vivo; however, in the presence of hyperlipidemia, there is reduced phagocytosis resulting in necrosis of apoptotic SMCs and leakage of intracellular IL-1. IL-1 acts on the surrounding viable SMCs and induces them to secret IL-6 and monocyte chemotactic protein (MCP)-1, thus initiating the progression of atherosclerosis [32]. This concept is further supported by many studies where aggressive treatment of hypercholesterolemia by statin therapy has resulted in marked reduction in the incidence of major cardiovascular event [33] (see Chap. 28).
Another feature of these early lesions of pathological intimal thickening is microcalcification, which has been demonstrated with anionic stains such as von Kossa’s or alizarin red stains. The apoptotic SMC remnants as well as calcium apatite crystals have also been demonstrated by transmission electron microscopy [34, 35]. The clinical significance of these early microcalcification lesions in lipid pools remains unknown; however, with time they likely coalesce along with calcifying macrophages and likely form calcified fragments.
25.5 Fibroatheroma
Fibroatheroma represents a further progressive stage of atherosclerotic disease and is identified by the presence of an acellular necrotic core, which can be distinguished from lipid pool lesions of PIT because they lack the presence of hyaluronan, proteoglycan such as versican and biglycan, and collagen (AHA type IV lesion) [1]. Fibroatheroma has been further classified by us into “early” and “late” necrotic cores, because we believe that this distinction may provide mechanistic insight into how necrotic cores form, grow, and evolve with time. The presence of “early necrotic core” is identified by the accumulation of macrophages within the lipid pool areas, and Bennett et al. have shown that apoptotic SMCs release IL-6 and MCP-1 which attract the macrophages and are involved in the breakdown of extracellular matrix, leading to the development of the necrotic core. Lesions with early necrotic core continue to show the presence of hyaluronan, versican, and biglycan and other matrix proteins focally, which are typically absent in more advanced “late necrotic core” fibroatheroma that exhibits necrosis, presumably due to release of matrix metalloproteinases by macrophages, resulting in complete breakdown of proteoglycans and collagen. Also, large numbers of macrophages within the areas of necrotic core display features consistent with apoptotic cell death, and free apoptotic bodies are abundant within the necrotic core, which has been aptly described by Ira Tabas as a “graveyard” of macrophages.
Increased accumulation of free cholesterol is another feature of late necrotic core. Acyl-coenzyme A:cholesterol acyltransferase 1, or ACAT1, esterifies free cholesterol and stores esterified cholesterol in macrophages [36]. Macrophage ACAT1 deficiency has been shown in mice to lead to an increase in atherosclerotic lesion area including free cholesterol accumulation in hyperlipidemic mice via disrupted cholesterol efflux, increased lipoprotein uptake, accumulation of intracellular vesicles, and accelerated apoptosis [37]. This together with the death of macrophages in the setting of defective phagocytic clearance of apoptotic bodies is thought to contribute to the development of late plaque necrosis [36, 38]. We have also shown that another source of free cholesterol in human necrotic cores is associated with the membrane of red blood cells, which are rich in free cholesterol (see below). The late necrotic core is devoid of any matrix and no longer stains for hyaluronan, proteoglycans, and collagen matrix as identified by specific stains for proteoglycans, hyaluronan, and Sirius red stain.
During the evolution toward an advanced fibroatheromatous lesion, there is an overlying layer of fibrous tissue (thick fibrous cap) composed mostly of type I and III collagen, proteoglycans, and interspersed SMCs, which play a critical role in harboring the contents of the necrotic core. The fibrous cap could undergo thinning and is now called a thin-cap fibroatheroma (TCFA) that may eventually rupture through mechanisms discussed below. The TCFA generally has a relatively large necrotic core with an overlying thin intact fibrous cap which is composed mainly of type I collagen with varying degrees of macrophages and lymphocytes. There is generally a paucity or absence of smooth muscle cells within the fibrous cap because of apoptosis. The fibrous cap thickness is a measure of plaque vulnerability, and its definition requires that it be ≤ 65 μm, which was based on the mean thickness of the thinnest part of the remnant cap of ruptured plaques that measured 23 ± 19 μm. Therefore, 95 % of the ruptured caps measured ≤ 65 μm [39].
25.6 Plaque Rupture
In regard to the characterization of thrombus composition, most acute thrombi at sites of rupture are platelet rich (“white” thrombi) (Fig. 25.3). Many factors including calcium, platelet surface receptors including GPIIb/IIIa or protease-activated receptors, and von Willebrand factor facilitate platelet aggregation and compaction of coagulum formation (Chap. 23). However, the proximal propagated thrombus exhibits mostly a fibrin-rich thrombus with interspersed red cells and is a red thrombus that occurs at sites of blood stasis because of absence of flow to the nearest side branch. The nature of the thrombus, i.e., red versus white thrombus, is of clinical importance, because it may be related to the effectiveness of thrombolytic therapy in patients presenting with acute myocardial infarction. Furthermore, the duration of the thrombus determines its characterization; a short duration forms a predominantly platelet-rich thrombus, while long-duration thrombi are likely to be longer in length and contain layered fibrin; further these thrombi begin to organize and are therefore more difficult to respond to thrombolytic therapy.
While the precise mechanism of plaque rupture is not fully understood, most researches agree that disruption of fibrous cap occurs due to the weakness of the cap from the presence of macrophages and T-lymphocytes. The macrophage infiltration is responsible for the collagen degradation and the lack of new collagen formation from interferon gamma, secreted by T-lymphocytes. Cap disruption results in exposure of necrotic core to the flowing blood, which activates coagulation and thrombus formation [1, 40]. It has only recently been appreciated that plaque ruptures are likely episodic and may occur in asymptomatic individuals [41] (Fig. 25.4). Moreover, at least 30 % of plaque ruptures occur at lesion sites with less than 75 % cross-sectional luminal narrowing [42]. We reported that the prevalence of coronary thrombosis at sites of insignificant narrowing (<70 % cross-sectional area narrowing) was 37 % in autopsy patients with plaque erosion and 26 % in plaque ruptures [42]. Mann and Davies reported that in plaques causing <50 % diameter stenosis, healed plaque rupture (HPR) was observed in <20 % of lesions, while in those with ≥51 % luminal stenosis, HPRs were observed in 73 % of plaques [43]. Also, we have similarly reported that 61 % of sudden coronary death victims have HPRs and were associated with healed myocardial infarction, increased heart weight, dyslipidemia, and diabetes mellitus [41]. Moreover, as the number of HPR in a specific lesion site increases, the degree of luminal narrowing is also progressing. Plaque ruptures without a previous healed rupture are unusual, i.e., virgin ruptures, and the latter are observed in only 11 % of patients dying suddenly [41].
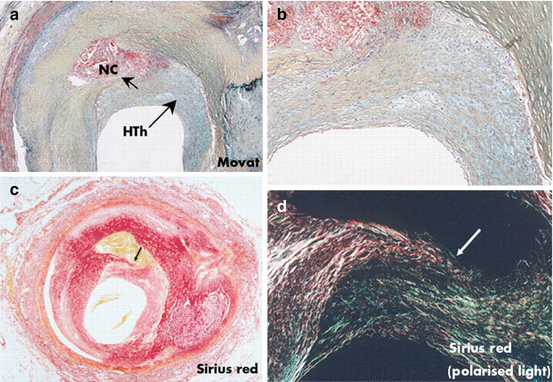
Fig. 25.4
Healed plaque rupture lesion with severe luminal narrowing. (a) Areas of intra-intimal lipid-rich core with hemorrhage and cholesterol clefts; an old area of necrosis (NC) is seen underlying a healed thrombus (HTh). (b) Higher magnification showing extensive smooth muscle cells within a collagenous proteoglycan-rich neointima (healed thrombus) with clear demarcation from the fibrous region of old plaque to right. (c, d) Layers of collagen by Sirius red staining. (c) Note area of dense, dark red collagen surrounding lipid hemorrhagic cores seen in corresponding view in (a). (d) Image taken with polarized light. Dense collagen (type I) that forms fibrous cap is lighter reddish yellow and is disrupted (arrow), with newer greenish type III collagen on right and above rupture site. (a, b) Movat pentachrome (Reproduced with permission from reference [42])
Recently, much interest has focused on the role of matrix metalloproteinases (MMP) as the main mechanism of fibrous cap disruption in coronary and carotid plaque ruptures. Type I collagen provides most of the tensile strength to the fibrous cap, and some pro-inflammatory cytokines, such as INF-γ, prevent collagen synthesis by smooth muscle cells [44]. The initial proteolytic breaks in collagen fibers I and III are caused by the presence of collagenases, such as MMP-1, MMP-8, and MMP-13, while the gelatinases MMP-2 and MMP-9 support the continued degradation of the interstitial collagens [40, 45–48]. However, the artery also has endogenous antagonists to MMPs, the tissue inhibitors of metalloproteinases (TIMPS) [49]. It has been demonstrated that “atheromatous” rather than “fibrous” carotid plaques preferentially express MMP-1 and MMP-13 [48]. Same authors also showed that collagenase-cleaved type I collagen occurs at sites that showed higher expression of MMP-1- and MMP-13-positive macrophages. There are other proteinases capable of degrading extracellular matrix including the cathepsin family (cathepsins S and K) and the inhibitor cystatin C [50]. However, these have strong elastolytic activity and have been implicated more with matrix remodeling and migration and proliferation of cells [46]. Although elastolysis may be more important in aneurysm formation, however, collagenolysis is the major determinant of plaque rupture [51]. While both collagenolysis and elastolysis are important components mediating plaque rupture, there are additional local factors contributing to this final event of arterial thrombosis, such as flow dynamics and vasospasm.
Apoptosis may also be important in the final development of plaque rupture [52]. Apoptosis of macrophages and SMCs has been observed in both progression and regression of atherosclerotic plaques [53]. Typically, the fibrous cap of plaque rupture sites shows very few SMCs, which are essential for synthesis of extracellular matrix proteins and maintenance of the fibrous cap [54]. Geng et al. in the mid-1990s demonstrated that mediators secreted by macrophages and T-lymphocytes including IFN-γ, FasL, TNF-α, IL-1, and reactive oxygen species accelerate smooth muscle cell apoptosis in vitro [55]. These upstream effectors can activate caspases causing mitochondrial dysfunction and death via the release of cytochrome c [52]. Apoptosis is considered to be the main mechanism of decrease in smooth muscle cells seen in thin-cap fibroatheroma and in ruptured plaques [31].
Today the “danger hypothesis” indicated that the endogenous self-adjuvants are released during cell death and are the inducers of inflammation that occur in the absence of infection and are referred to as damage-associated molecular patterns (DAMPs) [56]. In atherosclerosis there are three main mechanisms that trigger inflammation: cholesterol crystals, oxidation-specific epitopes, and IL-1α. Apoptotic SMCs are phagocytosed by adjacent SMCs; however, in the presence of hyperlipidemia, there is reduced capacity for phagocytosis by SMCs that results in necrosis of apoptotic SMCs [32]. Necrotic VSMCs release IL-1α, which activates adjacent viable VSMCs to produce the pro-inflammatory cytokines IL-6 and monocyte chemotactic protein 1 [32].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


