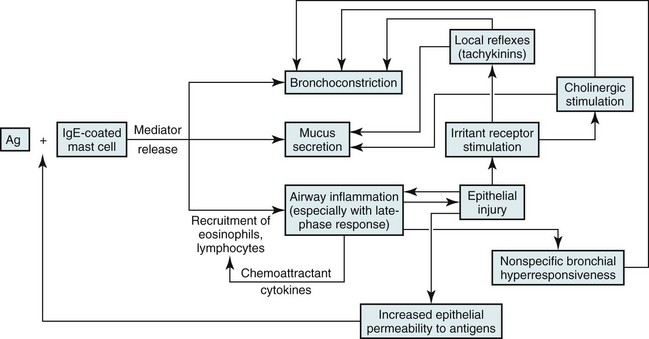
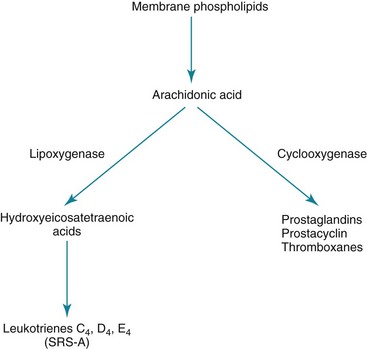
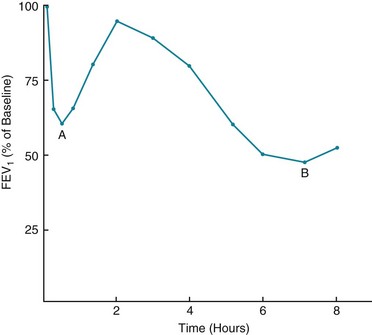
5 Chapter 4 discussed the normal structure of airways and considered several aspects of airway function. The most common disorders disrupting the normal structure and function of the airways—asthma and chronic obstructive pulmonary disease—are discussed here and in Chapter 6, respectively. Several other miscellaneous diseases affecting airways are covered in Chapter 7. No single factor or cell appears to be responsible for asthma; rather, a complex and interrelated series of events, including cellular infiltration, cytokine release, and airway remodeling, likely culminates in airway hyperresponsiveness and episodes of airflow obstruction (Fig. 5-1). A variety of mediators released from inflammatory cells can alter the extracellular milieu of bronchial smooth muscle, increasing its responsiveness to bronchoconstrictive stimuli. Mediators that have been proposed to play such a role include prostaglandin and leukotriene products of arachidonic acid metabolism. Some cytokine mediators released from inflammatory cells have various effects on other inflammatory cells, thus perpetuating the inflammatory response. For example, lymphocytes of the TH2 phenotype, which are thought to be a prominent component of the inflammatory response in asthma, release interleukin (IL)-5, which has a chemoattractant effect for eosinophils. IL-5 also stimulates growth, activation, and degranulation of eosinophils. IL-4, another cytokine released from TH2 lymphocytes, exerts a different type of proinflammatory effect by activating B lymphocytes, enhancing synthesis of IgE and promoting differentiation of TH2 cells. Mediators released from inflammatory cells may produce tissue damage that contributes to asthma pathogenesis. For example, when eosinophils degranulate, they release several toxic proteins from their granules, such as major basic protein and eosinophil cationic protein. These and other eosinophil products may contribute to the epithelial damage found in the asthmatic airway. Once the epithelium is injured or denuded, its barrier function is disrupted, allowing access of inhaled material to deeper layers of the mucosa. Additionally, the epithelial cells themselves may become actively involved in amplifying the inflammatory process (through production of cytokine and chemokine mediators) and in perpetuating airway edema (through vasodilation mediated by release of nitric oxide, leukotrienes, and prostaglandins). Finally, sensory nerve endings in the airway epithelial layer may become exposed, triggering a reflex arc and release of tachykinin mediators (e.g., substance P, neurokinin A), as shown in pathway 4 of Figure 4-3. These peptide mediators, released at bronchial smooth muscle, submucosal glands, and blood vessels, are capable of causing bronchoconstriction and airway edema. The pathogenetic mechanisms leading to bronchoconstriction are best defined for allergen-induced asthma. Allergens to which an asthmatic person may be sensitized are widespread throughout nature. Although patients and clinicians often first consider seasonal outdoor allergens such as pollen, many indoor allergens may play a more critical role. These allergens include antigens from house dust mites (Dermatophagoides and others), domestic animals, and cockroaches. Inhaled antigens are initially identified and processed by antigen-presenting cells called dendritic cells, which in turn present the antigenic material to T lymphocytes. Chemicals released by TH2 cells, especially IL-4 and IL-13, signal B lymphocytes to produce antigen-specific IgE antibodies. When an asthmatic person has IgE antibody against a particular antigen, the antibody binds to high-affinity IgE receptors on the surface of tissue mast cells and circulating basophils (see Fig. 5-1). If that particular antigen is inhaled, it binds to and cross-links IgE antibody (against the antigen) bound to the surface of mast cells in the bronchial lumen. The mast cell then is activated, leading to release of both preformed and newly synthesized mediators. Mediators released from the mast cell induce bronchoconstriction and increase airway epithelial permeability, allowing the antigen access to the much larger population of specific IgE-containing mast cells deeper within the epithelium. Binding of antigen to antibody on this larger population of mast cells again initiates a sequence of events leading to release of chemical mediators capable of inducing bronchoconstriction and inflammation. Several mediators have been recognized (Table 5-1), but the discussion here is limited to the few that have been primarily implicated in the pathogenesis of allergic asthma; major mediators include histamine and leukotrienes. Histamine: This relatively small (molecular weight 111) compound is found preformed within the mast cell and is released on exposure to the appropriate antigen. Histamine has several effects that may be important in asthma, including contraction of bronchial smooth muscle, augmentation of vascular permeability with formation of airway edema, and stimulation of irritant receptors (that can trigger a reflex neurogenic pathway via the vagus nerve, causing secondary bronchoconstriction). Despite these varied effects, the fact that the clinical manifestations of asthma do not respond to antihistamines suggests that histamine is not the most important chemical mediator involved. Leukotrienes: The leukotrienes include a series of compounds (LTC4, LTD4, and LTE4) that formerly were called slow-reacting substance of anaphylaxis (SRS-A). Unlike histamine, leukotrienes are not preformed in the mast cell but synthesized after antigen exposure and then released. To some extent, their actions are similar to those of histamine; they also have a direct bronchoconstrictor action on smooth muscle, increase vascular permeability, and stimulate excess production of airway mucus. Leukotrienes are synthesized from arachidonic acid (also the precursor for prostaglandins) but along a different pathway involving a lipoxygenase enzyme, as opposed to the cyclooxygenase enzyme used for prostaglandin synthesis (Fig. 5-2). LTC4 and LTD4 in particular are extraordinarily potent bronchoconstrictors and may have an important role in the pathogenesis of bronchial asthma. An interesting sidelight is provided by knowledge that some persons with asthma experience exacerbations of their disease after taking aspirin or other nonsteroidal antiinflammatory drugs (NSAIDs). These drugs are known inhibitors of the cyclooxygenase enzyme and may result in preferential shifting of the pathway shown in Figure 5-2 toward production of the bronchoconstrictor leukotrienes. The role of other mediators listed in Table 5-1 in asthma pathogenesis is less clear. Platelet-activating factor has been proposed to play a role in recruiting eosinophils to the lung, and platelet-activating factor activates eosinophils, stimulating them to release proteins toxic to airway epithelial cells. Late-Phase Asthmatic Response: The airway response to antigen challenge, as measured by changes in forced expiratory volume in 1 second (FEV1), appears to be more complicated and involves more than just the rapid mediator-induced bronchoconstriction seen within the first half hour following exposure. In many patients, the return of FEV1 to normal is followed by a secondary delayed fall in FEV1 occurring hours after antigen exposure (Fig. 5-3). This delayed fall in FEV1 is accompanied histologically by inflammatory changes in the airway wall. At the same time, increased bronchial hyperresponsiveness to nonspecific stimuli, such as histamine or methacholine, can be demonstrated and can last for days.
Asthma
Etiology and Pathogenesis
Airway Inflammation and Bronchial Hyperresponsiveness
Common Provocative Stimuli
Allergen Exposure
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Asthma