Location
Procedure
Restenosis (%)
References
Coronary
PTA
21–55
Coronary
BMS
16–44
Coronary
DES
10
Coronary
Atherectomy
27–39
Renal
PTA
26–100
[14]
Renal
BMS
17–60
Renal
DES
18
[16]
Iliac
PTA
22–34
[16]
Iliac
BMS
10–26
[16]
Femoral popliteal
PTA
39–62
Femoral popliteal
BMS
34–47
Femoral popliteal
DES
44
[21]
Femoral popliteal
Covered stent
37
[22]
Carotid
Endarterectomy
1.7–9.3
Carotid
BMS
2.7–21
Mesenteric celiac
PTA
36–64
Mesenteric celiac
BMS
40
Parallel advances in device technology also met with mixed results. The first major breakthrough came in the late 1980s with the introduction of the bare metal stent (BMS). BMSs countered elastic recoil and vasospasm that acutely decrease lumen diameter after angioplasty by 10–30 % [1, 2]. Stents were designed to hold lumen dimensions to those achieved at peak balloon inflation by providing radial strength in excess of inward radial forces exerted by the surrounding vessel wall. This strategy worked, but the greatest impact of BMSs has subsequently been recognized to be prevention of chronic arterial wall recoil, known as geometric remodeling of the healing vessel wall, as will be discussed below. Landmark PCI trials comparing BMSs to angioplasty reduced absolute and relative restenosis by 10 and 30 %, respectively [1, 2]. BMSs also improved PCI for chronic total occlusions, vein graft stenosis, and acute myocardial infarction. Nonetheless, restensosis after BMS remains unacceptably high at 15–32 % [1, 2].
Another decade passed before the next major advance, a period when exciting new approaches were embraced only to fade after disappointing results in randomized trials (e.g., atherectomy, gene therapy, and brachytherapy) [12, 13]. Drug-eluting stents (DESs) then emerged with startling results in phase I trials, reporting near 0 % restenosis with sirolimus-eluting stents [9]. Subsequent trials in less selective “real-world” populations have yielded slightly less favorable results in PCI, but after a decade of iterative refinement (e.g., new drugs and stent coatings), restenosis after DES is in the range of 5–15 %, a significant improvement over BMSs [4]. Other trials have showed that DESs are superior to angioplasty in treating the difficult problem of in-stent restenosis [35].
Restenosis in extracoronary vascular beds has received more attention in recent years, and rightly so, as the numbers of failures at these sites can be remarkably high, particularly for atherosclerotic lesions in the extremities (Table 14.1). For example, carotid revascularization carries a relatively low (∼6 %) risk of clinically significant restenosis (>70 % diameter stenosis) for both endarterectomy and stenting according to recent data from CREST (Lal et al. [36] International Stroke Conference 2012, New Orleans, LA). Much higher rates have been reported in endarterectomy trials using 50 % stenosis as the criteria for failure. Restenosis after renal angioplasty for atherosclerosis is so common that virtually all renal interventions done now employ primary stenting [14]. A meta-analysis of renal stenting by Leertouwer [14] found a mean rate of in-stent restenosis of 17 %, but late failures are probably much more common (Table 14.1). Large prospective renal stent trials have recently been completed, so the durability of BMSs will soon be characterized more accurately (e.g., ASTRAL and CORAL).
Treatment of infrainguinal peripheral artery disease is rapidly expanding as devices specifically tailored to the extremities are being developed and public awareness is increasing. However, the promise of less invasive options to replace high-risk open surgery has been slowed by rates of restenosis that often exceed 50 % at 2 years (Table 14.1). Moreover, the gains for the BMS and DES achieved in PCI have been far less consistent beyond the first few months in the lower extremities (Table 14.1) [37].
Incremental improvements from better patient and lesion selection, enhanced anticoagulation and anti-platelet agents, and improved devices and imaging have each helped reduce restenosis, but despite these advances, millions of persons are affected worldwide at an estimated cost for repeat interventions measured in the billions of healthcare dollars [38].
Structural Basis of Recurrent Lumen Narrowing
Despite the steady push to reduce the invasiveness of vascular reconstruction, virtually all methods are still mechanical in nature and cause trauma to the treated vessel wall, no matter how carefully applied. Surgery (bypass and endarterectomy) and endovascular approaches (angioplasty, atherectomy, and stenting) all injure the vessel wall, and the response to injury and subsequent healing are the common thread in the pathogenesis of restenosis.
Vessel wall trauma creates a thrombogenic lumen surface by denuding the protective endothelium, fracturing inelastic atherosclerotic plaque, and often wounding deep into the surrounding tunica media and adventitia (Fig. 14.1). This induces acute platelet and leukocyte adhesion and direct destruction of resident smooth muscle cells (SMC) and extracellular matrix (ECM), setting into motion a proliferative wound-healing response (termed “intimal hyperplasia”) that is generally proportional to the extent of injury. Accumulation of new cells and ECM at sites of injury forms a “neointima” layer at the vessel lumen surface, a feature common to virtually all forms of reconstruction and one of two major factors contributing to recurrent lumen encroachment (Figs. 14.1, 14.2, and 14.3).
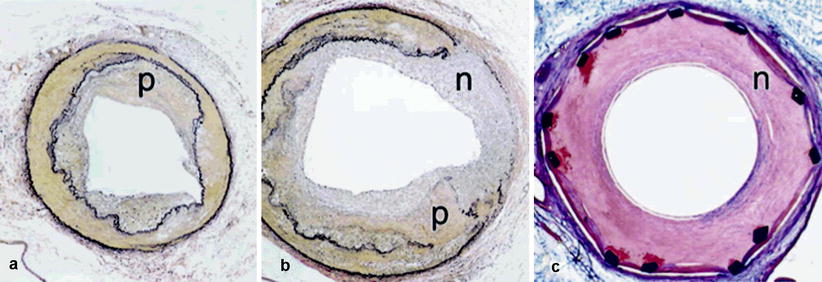
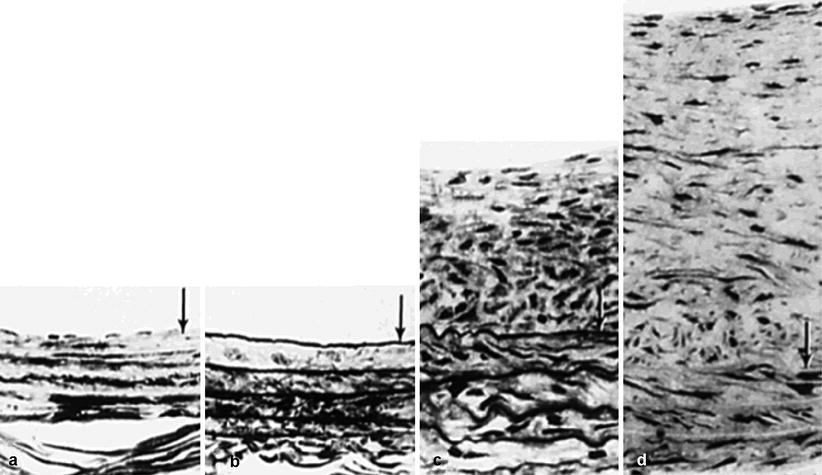
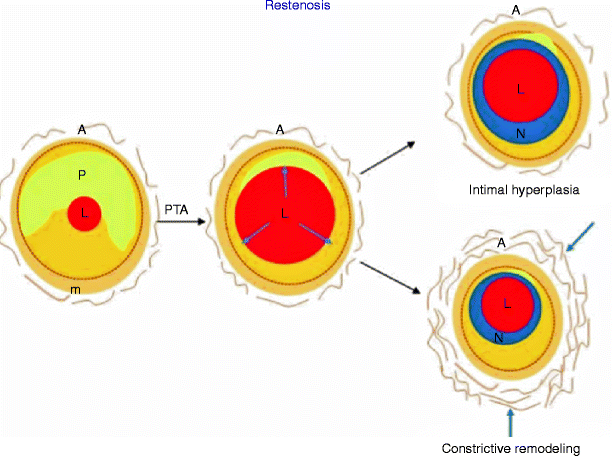
Fig. 14.1
Composite photomicrograph demonstrates a typical response 4 weeks after angioplasty and stenting in atherosclerotic primate. (a, b) Show the uninjured and injured common iliac arteries, respectively. Panel c shows stented iliac artery. Animals consumed an atherogenic diet for 2.5 years to create complicated plaques (p). (b) Shows fractured plaque (p) and overlying media that have healed with neointimal ingrowth (n). (c) Shows a stented monkey iliac artery with a typical neointimal lesion (n). (a, b) Verhoeff–van Gieson’s stain; (c) trichrome stain; original magnification all panels, ×40 (Reproduced by permission of American Heart Association, Cherr et al. [73])
Fig. 14.2
Composite photomicrograph of the rat carotid after balloon injury. Histological cross-sections of normal rat carotid artery before injury (a), immediately after balloon injury (b), after 2 weeks (c), then 12 weeks (d). Note significant intimal hyperplasia at 2 and 12 weeks with increasing contribution from ECM. Lumen is at top. Arrows indicate IEL (Reproduced with permission from [44])
Fig. 14.3
Mechanisms of lumen narrowing after angioplasty. Diagram illustrates the early concept of restenosis following angioplasty due entirely to neointimal formation (top) and more contemporary model, which includes both intimal hyperplasia and constrictive remodeling (bottom). L lumen, P plaque, A adventitia, N neointima
This observation led to an intense focus on the molecular and cellular regulation of vascular SMC growth and migration and ECM production and regrowth of a protective endothelial cell (EC) monolayer. Basic restenosis research initially focused on vein graft arterialization and prosthetic graft anastomotic intimal hyperplasia and then intensely on neointima formation at sites of angioplasty and within BMS. Animal models of angioplasty and bypass procedures were developed (Figs. 14.1 and 14.2) and the time course of intimal hyperplasia carefully described.
Potent inhibitors of experimental neointima formation were identified, but a series of clinical trials based on these results failed miserably with no impact on restenosis. This caused researchers and funding agencies to take a step back in the early 1990s to explore possibilities for the failure of animal models to predict clinical benefit. Three hypotheses emerged from this reappraisal. The first was the possibility that species differences existed in the regulation of SMC growth, so that drugs effective in rodents were ineffective in human beings. Data emerged to support this hypothesis as results from studies in nonhuman primate models of restenosis mirrored those in clinical trials [39]. The second hypothesis was that animal models did not accurately depict the background of complex advanced atherosclerosis and its contribution to the injury response. Such lesions are present in the vast majority of vessels treated clinically. To this day, few models come close to developing plaques that mirror the complexity of human lesions. Perhaps the closest approximation is the monkey model of diet-induced atherosclerosis [40]. When fed an atherogenic Western diet for 2–5 years, macaques develop complex lesions in the coronary, aortic, and iliac arteries (Fig. 14.1), but the expense and time required to work in this model have severely restricted its application. The third and perhaps most important hypothesis was that factors other than neointima were contributing to lumen loss after angioplasty, and confirmatory data soon emerged from BMS and IVUS trials. The concept of geometric arterial wall remodeling after angioplasty had been largely overlooked until serial imaging of human coronary lesions with IVUS demonstrated that changes in wall diameter were even more important than new wall mass (neointima) in the loss of lumen caliber over time [41–43]. We now understand that the combination of neointima formation and inward shrinkage of the vessel wall merge to produce lumen narrowing at sites of angioplasty in the absence of a stent (Fig. 14.3). While these two processes occur together, conceptually it is easier to consider their pathogenesis separately.
Intimal Hyperplasia
In practice, angioplasty is applied to stenoses surrounded by advanced atherosclerotic plaques. These lesions are inelastic, in contrast to the normal arterial wall, and rupture rather than stretch when dilated (Fig. 14.1) [39, 40]. New tissue formation following the procedure is termed “neointima” as it accumulates at the interface between the damaged vessel wall and flowing blood (Fig. 14.1). As noted above, intimal hyperplasia is a generic response to virtually all forms of mechanical arterial wall injury (e.g., angioplasty, atherectomy, endarterectomy, stenting, etc.). The time course and regulation of intimal hyperplasia following artery injury have been extensively characterized in the rat carotid balloon injury model by Clowes and others (Fig. 14.2) [44, 45].
Balloon injury denudes the endothelium and damages the surrounding wall by stretching, which if severe can rupture the tunica media. In the rat model, a stretch injury standardized to not rupture the IEL results in the immediate death of 25–30 % of medial SMCs. The loss of an endothelial barrier promotes platelet and leukocyte adhesion forming a thin layer of mural thrombus and an acute wave of inflammation. Adherent platelets release factors that promote cell growth and migration, including PDGFs, EGF, basic FGF, and TGF-β. Dead and injured SMCs release intracellular stores of basic FGF, and the damaged ECM releases matrix-bound growth factors as well as matrix fragments that have endogenous mitogenic and inflammatory properties. A dramatic wave of replication ensues in which 30 % of medial SMCs enter S-phase within 24 h. By day 4, both quiescent and replicating SMCs can be seen migrating from the media across the IEL to form a neointima (Fig. 14.2) [44–46]. Cells arriving in the neointima continue replicating until a quiescent state is reached by 4 weeks. The neointima continues to expand for ∼6 weeks from accumulation of additional ECM, and at 3 months ECM accounts for almost 80 % of the neointimal area in the rat model (Fig. 14.2d) [44].
In the rat carotid balloon injury model, the endothelium does not completely regenerate, but in other models, regrowth of an intact endothelial monolayer serves to shut down replication of the underlying SMC replication. This is due to paracrine effects of growth inhibitors secreted by the endothelium, including nitric oxide (NO) [47] and heparan sulfate proteoglycans [48]. Endothelium also serves a barrier function and excludes platelets, clotting proteins, and leukocytes from continuing to stimulate adjacent SMCs with their mitogenic, motogenic, and proinflammatory effects. These facts have formed the basis of clinical trials designed to enhance early endothelialization of prosthetic bypass grafts, BMS, and sites of angioplasty [49, 50].
As noted above, many drugs have been identified that inhibit intimal hyperplasia in the rat and other animal models of vascular injury, including heparin [48], angiotensin-converting enzyme inhibitors [51, 52], and NO donors, but when applied in human angioplasty restenosis trials, their effects have all been disappointing [32, 33]. This is due in part to the fact that intimal hyperplasia (new wall mass) is only one variable in the angioplasty restenosis equation, the other being constrictive arterial wall remodeling.
Arterial Wall Remodeling
Remodeling refers to changes in the arterial wall geometry (intima, media, and adventitia) that can occur independent of a change in the wall cross-sectional area. Normal adaptive vascular remodeling occurs when the vessel wall dilates and thickens in response to chronic increases in flow and pressure, respectively. Compensatory outward remodeling is a well-established response through which the media expands to accommodate the accumulation of atherosclerotic plaque, as described by Glagov and others. After angioplasty, the arterial wall seldom dilates to a greater caliber than was achieved when the balloon was inflated, but instead various amounts of inward wall shrinkage occur over time. As noted above, acute recoil and spasm decreases wall caliber by 10–30 % within minutes. Over the next few weeks, another constrictive process remodels the arterial wall to an even smaller caliber through a process with many parallels to wound contraction in healing cutaneous wounds.
If the reader has trouble visualizing the effects of remodeling, the data from BMS studies may be helpful. When a BMS is used, the amount of neointma that forms within the stent is generally worse, with a greater cross-sectional area than after angioplasty alone. Even so, stents reduce the frequency of restenosis compared to angioplasty alone because they completely block acute and chronic recoil/remodeling/shrinkage of the arterial wall. So the initial lumen caliber (the stent caliber) is large enough that moderate amounts of neointima have minimal impact on the lumen cross-sectional area. In contrast, an artery that constricts after angioplasty, remodeling pre-existing media and plaque area into a slightly smaller diameter, will severely narrow the lumen even if the area of the wall stays constant (i.e., no additional neointima, Fig. 14.3). These concepts have been confirmed in studies using serial imaging of arterial segments after angioplasty and after BMS. IVUS of the cross-sectional vessel area shows that after angioplasty 60–80 % of lumen loss was due to constrictive remodeling of the wall caliber, while only 20–40 % was from a change in total wall cross-sectional area (neointima) [41–43].
Changes in wall caliber are generally measured histologically by the area encompassed by the EEL [53]. The molecular mechanisms of constrictive reorganization of cells and ECM material in the injured arterial wall remain poorly understood, but attempts to explain it have drawn parallels to the process of cutaneous wound healing, wound contraction, and fibrosis [54–57]. In cutaneous wounds left to heal by secondary intent, the sequence of cellular events is similar in many ways to healing of the arterial wall after angioplasty. A platelet and fibrin plug forms in the defect, which serves as provisional matrix for the influx of leukocytes then fibroblasts from damaged wound edges as well as ingrowth of new microvessels. Wound fibroblasts dedifferentiate into myofibroblasts, which like SMCs express α-actin and are capable of contracting in response to vasoconstrictors [56]. As leukocytes remove clot and wound debris, myofibroblasts proliferate and lay down new ECM rich in glycosaminoglycans and proteoglycans to form granulation tissue [57]. Fibroblasts then reorganize and apply traction to the newly synthesized matrix, and wound contraction occurs to bring wound edges together. Subsequent ECM turnover leads to a change in matrix composition to a more collagen-rich material. This results in fibrosis and eventually a mature scar after many months [55].
Parallels occur in atherosclerotic arteries injured by angioplasty [54]. This can be modeled in nonhuman primates with complex pre-existing atherosclerosis. In this model, species differences in the regulation of SMC growth are minimized, and complex atherosclerotic lesions more closely depict the human plaque prompting reconstruction (Fig. 14.1). Angioplasty increases lumen size by fracturing the atherosclerotic plaque and stretching or tearing the surrounding media and adventitia. Thus, the arterial wall “wound” is transmural. The healing response is also transmural, and while new tissue is termed the neointima, it can develop in the media and adventitial layers if the fracture is deep.
Damage leads to acute inflammation where thrombus forms within fractures, dissections, and clefts, providing provisional scaffolding for the influx of leukocytes, initially neutrophils then monocytes after days 2–7 [40, 54]. Platelets and the fibrin plug attract leukocytes to the denuded area through a cascade of adhesion molecules including P-selectin, integrin Mac-1 and glycoprotein IIb/IIIa, and direct leukocyte attachment and migration [58, 59]. Mac-1 and platelet-mediated leukocyte adhesion plays an important role in vascular inflammation and restenosis. Following a brief but intense wave of proliferation, peaking at 4 days, SMC and other actin-positive cells (e.g., adventitial fibroblasts and circulating progenitor-derived SMCs) migrate to the lumen interface into the area of injury to form neointima starting at day 7. Inflammation then resolves, and neointima replaces the thrombus and thickens substantially from further accumulation of SMCs and ECM from days 14 to 28.
Constrictive remodeling of the arterial wall, probably from adhesive tension generated in reorganization of newly synthesized ECM, is often remarkable during this time period [53, 54]. Despite the accumulation of neointima, restenosis in this model is not caused by increased lesion size alone. Rather, the change in overall arterial wall size or remodeling with wall shrinkage is the major factor leading to lumen narrowing. Given that matrix accounts for more than 60 % of arterial wall volume, matrix reorganization is a key factor in wall remodeling and dependent on cell-matrix adhesive interactions, matrix degradation by proteases, and new matrix synthesis.
As in healing wounds, the composition of the new ECM at sites of vascular injury is distinct from that in normal arterial wall [54, 60–64]. The uninjured media is rich in types I and III collagen and elastin with lesser amounts of glycosaminoglycans and proteoglycans [61]. In contrast, ECM deposited by neointimal cells is rich in hyaluronan (HA) and versican. HA is a large hydrophilic disaccharide polymer associated with tissue remodeling in embryogenesis, wound healing, and tumor invasion. HA is important for scarless fetal wound healing and improves collagen remodeling and re-epithelialization when applied exogenously to cutaneous wound in adults [65–67]. Versican is a proteoglycan that binds to the HA backbone and changes its physical-chemical properties, increasing viscoelasticity, and together they provide hydrated space favorable for cell motility, invasion, and replication [68]. The relative ratio of HA and other stuctural matrix proteins such as collagen and elastin alters ECM reorganization in culture and within the healing arterial wall [62, 69].
HA is expressed in all arterial wall layers after angioplasty, most prominently the adventitia and neointima [54, 64]. Co-localization with type-I collagen in the healing arterial wall suggests an important interaction supported by in vitro studies of collagen remodeling. Adventitial fibroblasts and medial SMCs cultured from monkey aorta contract collagen lattices using β1 integrins. Addition of HA significantly enhances collagen reorganization and peri-cellular collagen fiber condensation [69]. This effect is mediated in part by cell surface receptors for HA, including RHAMM and CD44. Both are upregulated by arterial injury [69, 70]. Thus, these data suggest HA could alter constrictive collagen remodeling in the healing arterial wall.
Matrix metalloproteinases (MMP) are important regulators of matrix degradation that are critical in SMC migration in vitro, but their roles in constrictive remodeling are less well established. Animal studies using MMP inhibitors showed conflicting results. In rat, rabbit, and pig models of arterial injury, MMP inhibition limited the injury response [71, 72]. In contrast, a continuous infusion of a broad-spectrum MMP inhibitor after angioplasty in atherosclerotic monkeys had no effect on arterial wall remodeling or hyperplasia [73]. Not surprisingly then, clinical trials of MMP inhibitors for PCI endpoints have been negative [74].
Reactive oxygen species (ROS) have also been implicated in constrictive remodeling. ROS are transiently increased across the arterial wall after angioplasty. In rabbits undergoing iliac balloon injury, superoxide dismutase (SOD) activity, which maintains the redox balance by catalyzing the reaction of O2– to H2O2, was decreased significantly as constrictive remodeling occurred [75]. Exogenous administration of SOD to the endothelium increased lumen caliber, inhibiting constrictive remodeling by 59 %. SOD also improved nitrate levels, suggesting improved remodeling was in part from increased NO production.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


