Fig. 14.1
ECG sinus rhythm (heart rate 56 bpm), first-degree atrioventricular block, anterior fascicular block, incomplete right ventricular block, characteristic epsilon waves in leads V1–V4, and also inverted T-waves in inferior lead and leads V2–V6, with prolonged QT interval (corrected QT–QTc = 463 ms). The right bundle is very abnormal with fractionation throughout the right bundle in keeping with a cardiomyopathic process
Echocardiogram revealed right ventricular enlargement with hypokinesis in the mid- and apical segments and mild tricuspid regurgitation. Also, right outflow dilatation (23 mm/m2) with normal systolic pulmonary pressure was observed. The left ventricle was mildly enlarged with an ejection fraction of 50 % without regional wall motion abnormalities.
In order to better evaluate the right ventricular size, motility, and ejection fraction, the patient underwent cardiac MRI that revealed right ventricular dilatation with wall thickening and moderate dyskinesia in the mid- and apical segments. Right ventricular ejection fraction was deeply reduced, nearly 35 %. Left ventricle was mildly dilated with normal ejection fraction (Fig. 14.2). Unfortunately, the patient refused gadolinium injection.
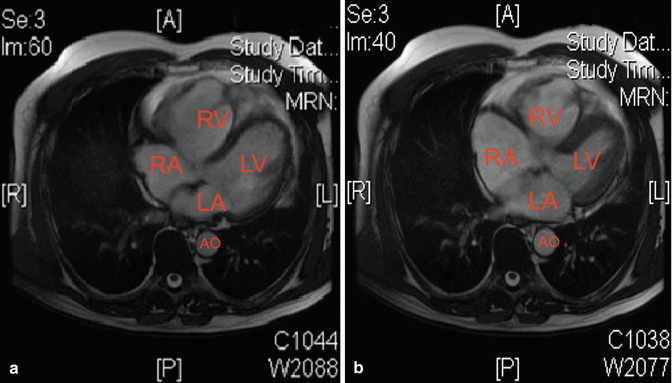
Fig. 14.2
Two axial FIESTA images (a, diastolic, and b, systolic) clearly depict a dilation of the four cardiac chambers, especially right ones. Wall of RV is prominently thinned
Clinical Course and Therapeutic Management
These imaging and clinical findings were strongly suggestive for ARVC.
According to the last AHA/ACC/ESC guidelines [1], taking into account the history of wide complex tachycardia suggestive for hemodynamically stable sustained VTs and severe right ventricle dysfunction, the patient was judged to be at moderate risk for SCD and underwent bicameral implantable cardioverter defibrillator (ICD) implant without major and minor complications. The decision to implant a bicameral ICD was based on the presence of first-degree AV block, anterior fascicular block, and incomplete right ventricular block, so the patient was judged to be at risk of developing high-degree AV block in the future. Sotalol was suspended and QTc interval became normal (440 ms). Therapy with a β-blocker was administered because of the recurrent sustained VTs. Also, a genetic test for ARVC-related mutations was performed.
On the 9th day, the patient was discharged on metoprolol tartrate 50 mg daily. Patient was advised to avoid competitive sport.
All the family members were recommended to undergo cardiologic visit, ECG and echocardiography evaluation, and eventually genetic screening once the mutation is identified in the patient.
14.2 Arrhythmogenic Right Ventricular Cardiomyopathy
The arrhythmogenic right ventricular cardiomyopathy (ARVC), also known as arrhythmogenic right ventricular dysplasia (ARVD), is a genetic disorder characterized clinically by malignant ventricular arrhythmias and an increased risk of sudden cardiac death (SCD), especially in young adults and athletes [2].
The ARVC is characterized by loss of myocytes and progressive fibroadipose replacement resulting in structural and functional changes of the right ventricle. However, considering the recent data showing a frequent involvement of the left ventricle, the ARVC is currently considered as a genetic disease of both ventricles and most properly called “arrhythmogenic cardiomyopathy” [2].
The adult prevalence in general population is about 1: 1,000–5,000 and is considered to be more common in individuals of Italian and Greek origin; however, the real prevalence in general population is likely to be underestimated [3].
Genetics
Data from genetic studies suggest that 30–50 % of cases are familial. The most common pattern of inheritance is an autosomal dominant form with incomplete penetrance and variable expressivity. The autosomal recessive inheritance form is less common and associated with cutaneous manifestations and wooly hair (Naxos disease, Carvajal syndrome).
To date, eight genes involved in the pathogenesis of CVAD have been identified; these genes encode for the desmosomal proteins [JUP (plakoglobin), DSP (desmoplachina), Pkp2 (placofilina2), Gene (desmoglein-2), Dsc2 (desmocollina-2)], the transforming growth factor (TGF β3), the transmembrane proteins (TMEM43), and the ryanodine receptor (RYR2)] [4].
Pathogenesis
The pathogenesis of ARVC is mostly related to reduced function of desmosomes which are intercellular adhesion complexes that provide mechanical connections between cardiac myocytes. In fact, it has been hypothesized that impaired cell adhesion may cause destruction and degeneration of cardiomyocytes, especially when subjected to mechanical stress (e.g., intense physical exercise) [2]. Because of the limited regeneration capacity of the myocardium, the repair process results in fibroadipose replacement which proceeds from the epicardium to endocardium. These process is completely absent at birth and likely begins during the puberty [2].
Anatomical fibrofatty replacement results in the free wall weakness and progressive ventricular dilatation with systolic dysfunction and aneurysm formation in the thinnest portions of the right ventricle, the so-called triangle of dysplasia (the apex, the inflow tract, and the outflow tract) [5]. Instead, fibrofatty replacement of the left ventricle generally involves the posterolateral wall, relatively thin, and rarely the interventricular septum [2].
Clinical Manifestations
The clinical presentation is more frequent between the second and the fourth decades. Because of genetic transmission with associated reduced penetrance, the severity of the clinical phenotype is somewhat variable.
In the natural history of ARVC, four stages are identified: (1) “concealed phase,” characterized by the absence of symptoms and minor structural abnormalities (the SCD can be the first and the only manifestation of the disease); (2) “overt electrical instability,” characterized by ventricular arrhythmias and morphofunctional cardiac abnormalities; (3) “right ventricular failure,” with severe systolic dysfunction of the right ventricle and initial or absent left ventricular abnormalities; and (4) “biventricular failure,” with severe systolic dysfunction of both ventricles similar to a cardiomyopathy [2].
Ventricular/Supraventricular Tachyarrhythmias and Sudden Cardiac Death
VTs in patients with ARVC range from single extra beats to complex VT, symptomatic and not, and the frequency appears to be proportional to the severity of the disease. The most common VT is generally monomorphic with origin from the right ventricle and left bundle branch block morphology. The arrhythmic episodes may origin from the apex, the inflow tract, or the outflow tract; when the site of origin is the right outflow tract, a differential diagnosis with idiopathic VT is required [2].
Unfortunately, the SCD may be the first manifestation of the disease. The estimated mortality rate for SCD varies from 0.08 to 9 % per year [1].
Exercise, especially the endurance sport, is considered a precipitating factor for arrhythmias in patients with ARVC. This “trigger” effect is likely related to the increased right ventricular stimulation by catecholamine exposure; in addition, data from literature suggest that the exercise itself contributes to the right ventricle dilatation and consequently to the disease progression [2].
Supraventricular tachyarrhythmia (SVT), such as atrial fibrillation, atrial tachycardia, and atrial flutter, in association with ventricular arrhythmias, is present in up to 25 % of patients with ARVC; less commonly, SVT may be the only arrhythmia present [6].
Left Ventricular Involvement
The wider use of cardiac MRI has allowed to appreciate a more common involvement of the left ventricle in ARVC [7].
In a study of 200 patients undergoing MRI, three patterns of disease expression were identified:
Classic: primarily affects the right ventricle and, only in advanced stages, the left ventricle (39 %).
Dominant left: characterized by early and severe involvement of the left ventricle and relatively mild disease of the right ventricle. It is very insidious variant as fibroadipose replacement involves initially only the epicardium of the left ventricle without causing wall motion abnormalities. The use of MRI contrast medium allows the identification of non-transmural scar.
Biventricular: characterized by a parallel involvement of both ventricles.
Diagnosis
The diagnosis of ARVC is complex and requires a high degree of clinical suspicion. To date, no single diagnostic test is enough sensitive and specific to confirm or rule out the disease; thus multiple parameters need to be considered.
The Task Force of the Working Group on Myocardial and Pericardial Disease of the European Society of Cardiology has developed the ARVC diagnostic criteria that take into account structural, histologic, arrhythmic, and genetic features. The original version of 1994 [8] that the revised version of 2010 [9] includes major and minor criteria collected in six main categories (Table 14.1). Based on this classification, definite diagnosis of ARVC requires two major criteria or one major and two minor or four minor criteria from different categories (Table 14.1).
2010 Task Force Criteria Definite = 2 major or 1 major + 2 minor or 4 minor from different categories Borderline = 1 major + 1 minor or 3 minor Possible = 1 major or 2 minor | |
1. Global/regional dysfunction/structural alterations | |
Major | By 2D echo: Regional RV akinesia, dyskinesia, or aneurysm and 1 of the following (end diastole): PLAX RVOT ≥32 mm (PLAX/BSA ≥19 mm/m2) PSAX RVOT ≥36 mm (PSAX/BSA ≥21 mm/m2) Fractional area change ≤33 % By MRI: Regional RV akinesia or dyskinesia or dyssynchronous RV contraction and 1 of the following: RVEDV/BSA ≥110 mL/m2 (male) or ≥100 mL/m2 (female) RV ejection fraction ≤40 % By RV angiography: Regional RV akinesia, dyskinesia, or aneurysm |
Minor | By 2D echo: Regional RV akinesia or dyskinesia and 1 of the following (end diastole): PLAX RVOT ≥29 to <32 mm (PLAX/BSA ≥16 to <19 mm/m2) PSAX RVOT ≥32 to <36 mm (PSAX/BSA ≥18 to <21 mm/m2) Fractional area change >33 to ≤40 % By MRI: Regional RV akinesia or dyskinesia or dyssynchronous RV contraction and 1 of the following: Ratio of RVEDV to BSA ≥100 to <110 mL/m2 (male) or ≥90 to <100 mL/m2 (fem) RV EF >40 to ≤45 % |
2. Tissue characterization of wall | |
Major | Residual myocytes <60 % by morphometric analysis (or <50 % if estimated), w/ fibrosis replacement of RV free wall myocardium in ≥1 sample, w/ or w/o fatty replacement of tissue on endomyocardial biopsy |
Minor | Residual myocytes 60–75 % by morphometric analysis (or 50–60 % if est.) w/ fibrous replacement of the RV free wall in ≥1 sample, w/ or w/o fatty replacement of tissue on endomyocardial biopsy |
3. Repolarization abnormalities | |
Major | TWI (V1, V2, V3) or beyond; >14 years of age; in the absence of complete RBBB QRS ≥120 ms |
Minor | TWI in V1 and V2; >14 years of age; in the absence of complete RBBB or in V4, V5, or V6 TWI in V1–V4; >14 years of age; in the presence of complete RBBB |
4. Depolarization conduction abnormalities | |
Major | Epsilon wave in right precordial leads (V1–V3) |
Minor | Late potentials by SAECG in ≥1 of 3 parameters in the absence of QRS duration of ≥110 ms on ECG: < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|