Group
Subgroup
1. Pulmonary arterial hypertension (PAH)
1.1. Idiopathic PAH
1.2. Heritable
1.2.1. BMPR2
1.2.2. ALK1, endoglin, SMAD9, CAV1, KCNK3
1.2.3. Unknown
1.3. Drug- and toxin-induced
1.4. Associated with
1.4.1. Connective tissue disease
1.4.2. HIV infection
1.4.3. Portal hypertension
1.4.4. Congenital heart diseases
1.4.5. Schistosomiasis
1′. Pulmonary veno-occlusive disease (PVOD) and/or pulmonary capillary hemangiomatosis (PCH)
1″. Persistent pulmonary hypertension of the newborn (PPHN)
2. Pulmonary hypertension owing to left heart disease
2.1. Left ventricular systolic dysfunction
2.2. Left ventricular diastolic dysfunction
2.3. Valvular disease
2.4. Congenital/acquired left heart inflow/outflow obstruction and congenital cardiomyopathies
3. Pulmonary hypertension owing to lung diseases and/or hypoxia
3.1. Chronic obstructive pulmonary disease
3.2. Interstitial lung disease
3.3. Other pulmonary diseases with mixed restrictive and obstructive pattern
3.4. Sleep-disordered breathing
3.5. Alveolar hypoventilation disorders
3.6. Chronic exposure to high altitude
3.7. Developmental lung disease
4. Chronic thromboembolic pulmonary hypertension (CTEPH)
5. Pulmonary hypertension with unclear multifactorial mechanisms
5.1. Hematologic disorders: myeloproliferative disorders, splenectomy, chronic hemolytic anemia
5.2. Systemic disorders: sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis
5.3. Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders
5.4. Others: tumoral obstruction, fibrosing mediastinitis, chronic renal failure segmental PH
Hemodynamic Basis of PH: An Introduction with Clinical Correlation
While a more detailed overview of pulmonary hemodynamics is presented in Chap. 10, a working knowledge of the hemodynamic basis of PH is extremely helpful in directing the clinical approach to the patient and so will be reviewed here in brief.
Current guidelines define PH as mean pulmonary artery pressure (mPAP) ≥25 mmHg with the patient at rest [6–8]. The definition of abnormal PAP response to exercise was considered as part of the 2013 WHO Nice deliberations, but no consensus was reached due to the lack of available data. Changes in pulmonary vascular and left ventricular compliance with aging make it difficult to determine normal and abnormal rises in PAP during exercise leading to the recommendation that exercise-induced PAP not be included in the definition of pulmonary hypertension [14]. It is crucial to note that while the pressure cutoff of 25 mmHg defines the presence or absence of disease by convention, it provides no information regarding its etiology. Application of Ohm’s law is used to describe the relationship between pressure and flow in the pulmonary circulation, but this assumes nonpulsatile, laminar flow within uniform, nondistensible vessels—all of which do not apply to the pulmonary vascular bed. With this limitation in mind, Ohm’s law dictates that mPAP is the product of pulmonary blood flow (Q p) and resistance across the pulmonary bed (pulmonary vascular resistance [PVR]), added to pulmonary venous pressure. In the absence of an anatomic shunt, Q p is approximately equal to systemic blood flow (Q s or cardiac output [CO]). Pulmonary venous pressure is difficult to measure directly in routine practice, so pulmonary capillary wedge pressure (PCWP) is used as a more readily obtained surrogate. Thus, an increase in mPAP may occur as a result of increases in Q p, PVR, or PCWP (Fig. 8.1). Each will be discussed in turn below.
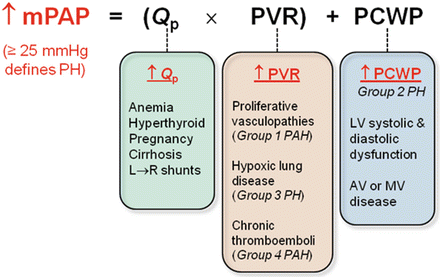
Fig. 8.1
Schematic representation of hemodynamic etiologies of increased pulmonary artery pressure. mPAP mean pulmonary arterial pressure, Q p pulmonary blood flow, PVR pulmonary vascular resistance, PCWP pulmonary capillary wedge pressure, LV left ventricle, PH pulmonary hypertension, PAH pulmonary arterial hypertension, AV aortic valve, MV mitral valve
Elevated Q p
Disorders associated with isolated elevations in pulmonary blood flow are relatively uncommon causes of pulmonary hypertension, since an otherwise healthy pulmonary vascular bed has the ability to recruit additional underperfused vasculature to accommodate increases in flow without a significant increase in pressure [15, 16]. In some circumstances, however, this reserve is impaired and elevations in Q p may drive, at least in part, a rise in mPAP. Usually, diagnosis of the primary condition predates the discovery of pulmonary hypertension, and there are frequently other symptoms that tend to dominate the clinical picture rather than the pulmonary hypertension per se. For these reasons, disorders associated with isolated increases in CO are not explicitly represented as a separate etiologic category in the current PH classification scheme [4].
Systemic conditions classically associated with an elevated CO include chronic anemia, pregnancy, hyperthyroidism, liver disease, and left-to-right shunts on the basis of either congenital heart disease, arteriovenous fistulae, or certain other disorders (e.g., Paget’s disease of bone, multiple myeloma). In anemia, elevated CO is a physiologic response aimed at maintaining adequate systemic oxygen delivery in the face of reduced oxygen carrying capacity. Modest levels of anemia commonly encountered in outpatient practice are not likely to cause significant elevations in CO, as a seminal hemodynamic study suggested a hemoglobin threshold of <7 g/dL is required to obligate a rise in CO in humans [17]. The increased metabolic demands of pregnancy imparted by the developing fetus also necessitate a rise in cardiac output, mediated by increases in both preload (due to plasma volume expansion) and heart rate and a reduction in systemic vascular resistance (SVR) [18]. Despite higher pulmonary blood flow, however, significant rises in PAP in healthy subjects typically do not occur presumably due to concomitant reductions in PVR [19]. Hyperthyroid states may be associated with elevated CO, wherein thyroid hormone can directly augment myocardial contractility as well as reduce SVR [20]. Interestingly, associations between abnormal thyroid function and PAH have been reported in up to half of such patients although hypothyroidism has been identified more commonly than hyperthyroidism [21, 22]. Thyroid disorders are particularly common in patients with APAH related to autoimmune disorders, potentially reflecting a common autoimmune basis in these patients. The potential pathogenic role of abnormal thyroid function in the development of PAH remains unclear; however, the association between thyroid dysfunction and pulmonary vascular disease is recognized by its inclusion in Group 5 (PH with unclear multifactorial mechanisms) of the current classification scheme [4].
Patients with end-stage renal failure who have an arteriovenous fistula for dialysis access are at risk for development of high cardiac output if the fistula flow becomes excessive. If such patients are undergoing hemodynamic catheterization, temporary occlusion of the fistula may enhance understanding of the contribution of the fistula to the high output state. Measurement of the fistula flow can be achieved by use of ultrasound or Fresenius clearance method [23]. If the fistula is resulting in excessive flow, banding of the fistula may be helpful. Arteriovenous malformations, particularly abdominal, also may result in high flow states.
Thus, while all these conditions can contribute to elevated cardiac output, they are rarely the sole cause of significant, symptomatic PH. However, they all may serve to unmask or exacerbate an underlying primary pulmonary vasculopathy (i.e., a process characterized by abnormal PVR) by superimposing an obligatorily increased Q p on an already restricted pulmonary vascular bed. In such a scenario, the normal plasticity of the pulmonary circulation is impaired which results in higher mPAP at any given level of flow.
In the context of a discussion on elevated pulmonary blood flow and PH, two conditions deserve additional comment. The first relates to elevations in Q p occurring in the context of a left-to-right anatomic shunt. Congenital defects in this category include ventricular septal defect (VSD), atrial septal defect (ASD), and patent ductus arteriosus (PDA), as well as more complex lesions such as truncus arteriosus, transposition of the great vessels, and syndromes characterized by single ventricle physiology. Anomalous pulmonary venous drainage (APVD) is another type of congenital defect associated with left-to-right shunting wherein one or more pulmonary veins return oxygenated blood to a systemic vein (most often the superior vena cava) or right atrium, and commonly co-exist with an ASD (usually sinus venosus type). In the case of VSD, ASD, or PDA, large defects typically present in infancy. However, smaller defects (in particular ASD) may go unnoticed for decades and not come to medical attention until well into adulthood [24, 25]. Initially, such anatomic defects cause PH by virtue of increased pulmonary blood flow mediated by the left-to-right shunt (elevated Q p:Q s), and PVR is normal. Over time, the increase in pulmonary blood flow can incite pathogenic changes within the pulmonary arterioles resulting in an obliterative pulmonary vasculopathy characterized by an elevated PVR. A higher pressure burden as seen in cases of VSD or PDA, combined with the detrimental effects of high flow and shear stress, may be an important factor in the more rapid development of pulmonary vascular disease in these subsets. As the process progresses, right-sided pressures increase, right ventricular insufficiency ensues, and the shunt may reverse (or become bidirectional) with resultant hypoxemia, known as Eisenmenger syndrome. Careful hemodynamic characterization with attention to the relative contributions of elevated Q p and/or PVR to elevated PAP and utilization of pulmonary vasodilator challenge to assess reversibility are crucial to determine whether correction of the defect and/or institution of pulmonary vasoactive therapy is warranted.
Cirrhosis of the liver, particularly when advanced, is also associated with a hyperdynamic state characterized by elevated systemic and pulmonary blood flow, reduced SVR [26], and potentially increased cardiac filling pressures due to excessive salt and water retention. When abnormal pulmonary pressures are identified in this population, it is more frequently related to elevated cardiac output and/or PCWP [27, 28]. However, similar to the case of an anatomic shunt, it is critical to distinguish this scenario from that where the primary hemodynamic abnormality is an elevation in PVR. This latter condition is known as portopulmonary hypertension (PoPH) and occurs in ~5 % of patients with chronic liver disease being evaluated for liver transplant [28, 29]. The presence of portal hypertension is a prerequisite for the development of PoPH, although there is no clear association between the etiology or severity of portal hypertension and the presence or severity of PoPH [30, 31]. PoPH is classified within Group 1 of the current PH classification scheme, displays pathologic changes identical to those seen in idiopathic PAH [32], and harbors a worse prognosis compared to patients with chronic liver disease without PoPH [33, 34]. Significant, untreated PoPH is associated with increased mortality in the setting of liver transplantation [33, 35, 36] and may represent a contraindication to the procedure. Patients being evaluated for liver transplantation should be screened for pulmonary hypertension by echocardiography and if found should be further evaluated by right heart catheterization [37].
Elevated PCWP
Elevation in left atrial pressure and subsequent pulmonary venous hypertension, as reflected by elevated PCWP, is the final common pathway by which various forms of left heart disease can cause PH. Systolic, restrictive, or infiltrative cardiomyopathies as well as mitral or aortic valvular disease all can lead to a rise in left atrial pressure that is transmitted back to the pulmonary arteries via the pulmonary veins. Heart failure with preserved ejection fraction (HFpEF) is increasingly recognized as a cause of chronic dyspnea, congestive heart failure, and PH. A community-based study of subjects ≥65 years of age suggests an overall prevalence of CHF of ~9 %, over half of which are associated with normal EF on echocardiography [38]. Comorbidities that frequently associate with HFpEF include older age, female sex, hypertension, atrial fibrillation, and obesity [39–41]. PH may be exceedingly common in elderly patients with HFpEF; a community-based echocardiographic study (wherein an RVSP ≥35 mmHg defined the presence of PH) has indicated an overall prevalence of PH in this population of 83 %, compared with 8 % in elderly hypertensive patients without HFpEF [42].
Occasionally, pulmonary venous hypertension may exist in the absence of left atrial hypertension in the setting of pathologic processes that primarily affect the pulmonary veins. Rarely, bulky adenopathy (such as that seen in sarcoidosis [43], tuberculosis [44], histoplasmosis, or other chronic granulomatous diseases or metastatic malignancy) or fibrosing mediastinitis [45] may lead to pulmonary vein obstruction. More recently, catheter ablation with pulmonary vein isolation, used for the treatment of atrial fibrillation, has been associated with the development of pulmonary vein stenosis and subsequent PH [46, 47].
Elevated PVR
Abnormalities in PVR may result from a broad array of pathophysiologic mechanisms, and thus, this hemodynamic abnormality encompasses a heterogeneous group of disorders spanning Groups 1, 3, 4, and 5 of the PH classification scheme. This chapter uses the term PAH (pulmonary arterial hypertension) to refer to conditions in this category wherein the primary pathophysiologic determinant is an elevated PVR.
Group 1 PAH
This group includes idiopathic (IPAH; formerly “primary pulmonary hypertension”), heritable (HPAH), and drug/toxin-mediated forms of pulmonary arterial hypertension as well as those associated with certain underlying conditions (APAH), namely congenital heart disease with left-to-right shunts, connective tissue disease (CTD), HIV infection, portal hypertension, and schistosomiasis infection. Two other rare forms of PH include pulmonary veno-occlusive disease (PVOD), characterized by obliterative lesions in the septal and preseptal pulmonary veins, and pulmonary capillary hemangiomatosis (PCH), characterized by extensive proliferation of the pulmonary capillaries into alveolar septa, bronchial walls, and the pleura; these entities are subclassified as Group 1′. PVOD may represent up to 10 % of PAH cases otherwise thought to be idiopathic [48]. It presents with symptoms similar to IPAH, but is associated with more severe derangements in oxygenation and diffusion capacity. PVOD can cause unique chest imaging findings not found in other forms of PH that should raise suspicion for this diagnosis (see section “Chest Computed Tomography (CT)”). Occult alveolar hemorrhage on bronchoalveolar lavage is also suggestive [49]. Patients with PVOD treated with pulmonary vasoactive therapy can develop life-threatening pulmonary edema due to pulmonary arteriolar vasodilation in the setting of fixed venous outflow obstruction [50].
All of the above entities are associated with similar pathologic findings within the pulmonary vascular bed, including medial hypertrophy, intimal and adventitial fibrosis, in situ thrombosis, and the advanced plexiform lesion [51]. As the disease process progresses and PVR rises, the afterload imposed on the right ventricle increases and higher pressures are required to maintain a given level of cardiac output.
Group 3 PH
Group 3 includes primarily respiratory-related disorders characterized by hypoxemia and/or hypoventilation, including all forms of obstructive and restrictive lung disease, neuromuscular or chest wall/skeletal disorders, and sleep-disordered breathing (obstructive sleep apnea and obesity hypoventilation syndrome). Patients in this group have been uniformly excluded from all of the pivotal, randomized, controlled trials of targeted PAH therapies. Although hypoxic vasoconstriction and subsequent vascular remodeling may be a final common pathway for the development of PH in many of these disparate conditions [52, 53] (discussed in greater detail in Chap. 4), other unique pathobiologic mechanisms exist that can contribute to elevations in PAP and PVR in each particular disorder. COPD, for example, is uniquely associated with hyperinflation, which can contribute to an elevated PVR due to increased alveolar pressure and compression of intra-alveolar vessels [54]. In idiopathic pulmonary fibrosis, there may be “spillover” of the inflammation in the epithelial compartment into the adjacent endothelial compartment, contributing to vascular obliteration and a rise in PVR [55, 56].
Group 4 PAH
Pulmonary arterial hypertension in this category is thought to occur from vascular obstruction due to recurrent embolic and/or in situ thrombotic events with organization and incorporation of the thrombotic material into the vessel wall. The incidence of chronic thromboembolic pulmonary hypertension (CTEPH) may be as high as 3.8 % up to 2 years after an embolic event [57]. However, because not all patients with CTEPH can recollect a discrete embolic event, the true incidence of the disease may be higher. While pulmonary arterial obstruction by organized thrombi is the sine qua non of CTEPH, patients can exhibit histopathologic changes in the small pulmonary arterioles that are indistinguishable from those seen in IPAH, and these have been observed to occur in regions of the lung spared from large-vessel obstruction [58].
Pulmonary thromboendarterectomy is a technically demanding procedure in which proximal organized thrombi are surgically removed. Individuals with more proximal, surgically accessible disease appear to derive the greatest benefit from the procedure [59], underscoring the importance of a meticulous characterization of the burden of pulmonary vascular disease and its location based on information from RHC, CT angiogram, pulmonary angiography, and, at some centers, pulmonary angioscopy [60]. Chapter 6 of this text presents a more in-depth discussion of CTEPH.
Group 5 PH
Group 5 represents a collection of disorders that have been associated with pulmonary arterial hypertension due to unique and/or ill-defined mechanisms. The current classification scheme subdivides such entities into specific categories, including hematologic (myeloproliferative disorders, splenectomy, chronic hemolytic anemias), systemic (sarcoidosis, pulmonary Langerhans cell histiocytosis, neurofibromatosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis), metabolic (glycogen storage disease, Gaucher disease, thyroid disorders), and other (tumor obstruction, fibrosing mediastinitis, chronic renal failure on dialysis).
Although some forms of Group 5 PH such as those associated with sarcoidosis and metabolic disorders closely resemble Group 1 PAH, every randomized controlled trial of PAH therapy has excluded patients with Group 5 disease.
Clinical Approach: History
A detailed history provides a wealth of information in the evaluation of a patient with known or suspected PH. Relevant elements of the history can provide information regarding the likelihood of PH being present, its potential cause or causes, and inform the clinician as to its severity and response to treatment.
The most common symptom associated with most forms of PH is dyspnea, particularly dyspnea on exertion. It is reported in virtually all patients and is often of a progressive nature. Other common symptoms include fatigue, exertional near-syncope or syncope, chest pain, edema, and palpitations [13]. The degree of dyspnea should be quantitated, and an assessment of functional class performed according to WHO criteria [61], modeled on the New York Heart Association functional class schema for patients with CHF (Table 8.2). Patients with PH may have a progressive decline in their functional capacity resulting in gradual curtailment of activities to a level where dyspnea is minimized. This can result in an apparent overestimation of functional ability by some patients, which can be elucidated with an assessment of specific activities and asking the patient how easily he or she can perform such activities now as compared with a point in the past (e.g., “How easy is it for you to climb two flights of stairs now as compared with 6 months or a year ago?” or “Are there any activities you used to do that you have stopped recently?”). Soliciting the perspective of a close contact can be helpful in corroborating or refining the patient’s assessment. Young patients, particularly those who are athletic or otherwise very fit, may report only a decrement in their ability to tolerate prolonged exercise which may be erroneously ascribed to deconditioning; this may in fact represent early pulmonary vascular disease with an impaired CO response to higher workloads.
Table 8.2
WHO functional class assessment for PH
Class I | Patients with PH but without resulting limitation of physical activity. Ordinary physical activity does not cause undue dyspnea or fatigue, chest pain, or near-syncope. |
Class II | Patients with PH resulting in slight limitation of physical activity. They are comfortable at rest. Ordinary physical activity causes undue dyspnea or fatigue, chest pain, or near-syncope. |
Class III | Patients with PH resulting in marked limitation of physical activity. They are comfortable at rest. Less than ordinary activity causes undue dyspnea or fatigue, chest pain, or near-syncope. |
Class IV | Patients with PH with inability to carry out any physical activity without symptoms. These patients manifest sights of right heart failure. Dyspnea and/or fatigue may even be present at rest. Discomfort is increased by any physical activity. |
The development of exertional chest discomfort or syncope is a potentially ominous sign of significant right ventricular (RV) ischemia and/or dysfunction and should impart an element of expediency to the diagnostic workup or modification of treatment in cases of known PAH. Anginal chest pain in PAH may arise due to increased oxygen demand from increased RV muscle mass, reduced oxygen supply due to reduced right coronary artery flow with impaired microcirculation due to right ventricular hypertrophy, and in some cases compression of the left main coronary artery by an enlarged pulmonary artery [62, 63]. In the seminal, prospective primary pulmonary hypertension registry, syncope eventually occurred in >1/3 of subjects [13] and is caused by the inability of the RV to increase CO in the face of increased demand and/or systemic vasodilation. Beyond the impairment to flow attributed to elevations in PVR, pathologic ventricular interaction results in reduced left ventricular diastolic compliance, which further limits the ability to maintain left ventricular stroke volume and adequate systemic perfusion pressure [64–67]. The abrupt onset of dyspnea in a patient with other clinical features to suggest PH should prompt consideration of acute pulmonary embolism (PE), either in isolation or superimposed on a background of chronic thromboembolic disease.
During the initial diagnostic phase, additional historical elements should be elicited to help formulate the differential diagnosis. A relatively short survey of comorbid conditions and possible associated symptoms will help the clinician quickly discern potential etiologic categories that may account for PH, and is summarized in Table 8.3. From a statistical perspective, given the relative rarity of idiopathic PAH, one should maintain a high index of suspicion of secondary pulmonary hypertension, keeping in mind that the most common cause of right heart failure is left heart failure. As such, consideration of various forms of left heart disease should be a priority in otherwise “undifferentiated” PH. Significant left ventricular systolic dysfunction or aortic or mitral valvular disease is usually readily apparent on transthoracic 2DE. An increasingly common clinical problem is that of differentiating pulmonary venous hypertension related to HFpEF (PH-HFpEF) and PAH. As discussed previously, PH appears to be a common occurrence in HFpEF [42]. Echocardiographic clues to the presence of HFpEF include Doppler features of significant diastolic filling abnormality and presence of left atrial enlargement. Conversely, some patients with PAH, particularly when more advanced, may display echocardiographic evidence of impaired left ventricular filling due to pathologic ventricular interaction [68]. In a cross-sectional study comparing PH-HFpEF and PAH, historical features more frequently associated with PH-HFpEF included older age and the presence of systemic hypertension or coronary artery disease [69]. (It should be mentioned that in this study, those with PH-HFpEF had some element of pulmonary vascular disease as the authors required an elevation in PVR or transpulmonary gradient as part of the definition of PH-HFpEF.) However, it is important to recognize that the presence of PAH is not protective against the development of these more common comorbidities and thus can co-exist. Moreover, while the classic presentation of IPAH is that of a young woman with progressive dyspnea, PAH is being diagnosed with increased frequency in the elderly; contemporary registries suggest that ≥20 % of PAH subjects are >60 years of age [10, 11].
Table 8.3
Historical elements and associated signs and symptoms pertinent to the evaluation of known or suspected pulmonary hypertension
Focused historical elements | Associated signs or symptoms | PH category |
|---|---|---|
Family history of pulmonary hypertension, sudden death, or ill-defined heart failure | Group 1 | |
Known congenital heart disease, protracted illness during infancy, “blue baby” | Cyanosis, polycythemia | |
Use of prescription weight loss pills (“fen-phen” or congeners), methamphetamine use | ||
Known or suspected HIV/AIDS, IV drug use | Generalized wasting, “track marks” | |
Liver disease or cirrhosis | Ascites, jaundice, encephalopathy, varices, or hemorrhoids | |
Travel to or residence in areas with endemic Schistosomiasis | Malnutrition/wasting, hepatosplenomegaly | |
Known or suspected connective tissue disease | Rash, telangiectasias, Raynaud’s phenomenon, photosensitivity, sclerodactyly, esophageal problems, oral ulcers | |
Valvular disease, coronary disease, cardiomyopathy, atrial fibrillation | Orthopnea, paroxysmal nocturnal dyspnea | Group 2 |
Smoking history, any “chronic lung problems,” occupational exposures, sleep disturbances | Chronic cough or sputum production, snoring, witnessed apneas | Group 3 |
Prior history of deep venous thrombosis or pulmonary embolism, hypercoagulable state | Asymmetric edema | Group 4 |
Myoproliferative disorder, splenectomy, chronic dialysis | Group 5 |
If suspicion of left heart disease as a primary cause of PH is relatively low, then a thorough search should be undertaken to identify other associated or secondary causes of PH. Connective tissue diseases are the most common underlying conditions predisposing to the development of PAH, and as such CTD features should be sought out. In a single-center cohort of elderly patients (>65 years of age) referred for evaluation of PH, the presence of CTD was found to be a strong predictor of PAH [70]. Scleroderma (both limited and diffuse forms) and mixed connective tissue disease are the two specific CTDs most commonly associated with PAH, followed by systemic lupus erythematosus (SLE), and less frequently rheumatoid arthritis, polymyositis/dermatomyositis, and Sjögren syndrome [11, 71, 72]. Occasionally, PAH may occur in the absence or precede the development of other symptoms or exam findings to suggest scleroderma (“scleroderma sine scleroderma”) or another specific CTD. A directed history aimed at symptoms of esophageal dysfunction, Raynaud’s phenomenon and autoantibody testing may help identify a “latent” CTD [73, 74]. A history of medical problems in the postnatal period or childhood heart murmur should be obtained as a clue to potential underlying congenital heart disease. The presence of chronic cough, sputum production, or wheezing may indicate underlying obstructive or restrictive lung disease. Snoring, witnessed apneas, morning headaches, or excessive daytime sleepiness suggests the presence of sleep-disordered breathing. A prior history of deep venous thrombosis or PE may raise the suspicion for CTEPH, although 25–50 % of CTEPH patients lack prior knowledge of any thrombotic event [75–77]. Large PE, unprovoked and/or multiple embolic events, younger age, or the presence of certain medical conditions, such as splenectomy, ventriculo-atrial shunts, inflammatory bowel disease, or positive antiphospholipid antibodies, may predispose to the development of CTEPH [57, 76, 78].
A social history should be performed, documenting any pertinent travel or occupational history, anorexigen (Aminorex or fenfluramine derivatives) or recreational (stimulants, including amphetamine compounds [79]) drug exposure, and risk factors for liver cirrhosis, HIV or viral hepatitis infection. While PAH associated with HIV infection remains relatively rare, the diagnosis should be considered in any patient with HIV with dyspnea of unknown origin, irrespective of the level of immune compromise [80]. A family history is necessary to assess for heritable PAH, which in a majority of cases is caused by mutations in the gene encoding the TGF-β receptor BMPR2 and displays an autosomal dominant pattern of inheritance [81–84]. However, due to incomplete penetrance, patients with heritable PAH may not always recollect a family member known to be afflicted with the disease. Alternatively, affected family members of previous generations may have been misdiagnosed as having another cardiopulmonary condition given a relative lack of awareness of PAH in preceding decades.
Clinical Approach: Physical Examination
In concert with a focused history, the physical examination can offer critical insights into the possible causes of PH and it severity. An anatomic-based approach is presented here.
Head and Neck
Underlying conditions that can predispose to PH often have manifestations in the head and neck. Sarcoidosis may be associated with cranial nerve deficits, parotid or lacrimal gland enlargement, ocular findings (uveitis, conjunctival nodules), or hypopigmentation around the hairline. Lymphadenopathy might suggest sarcoidosis or a lymphoproliferative disorder. Scleral icterus suggests hepatobiliary dysfunction. Proptosis in conjunction with an enlarged thyroid may be indicative of Graves’ disease. An enlarged tongue, or periorbital purpura, may suggest systemic amyloidosis. Stigmata of connective tissue disease include malar rash, oral ulcers, loss of naso-labial folds, microstomia, and telangiectasias. A preponderance of telangiectasias involving mucous membranes should also raise suspicion of hereditary hemorrhagic telangiectasia (Osler-Weber-Rendu syndrome). A short, thick neck with low-riding soft palate or tonsillar crowding should raise the possibility of obstructive sleep apnea. Hypoxemic patients may display peri-oral cyanosis. Ulceration of the nasal septum may be seen in either sarcoidosis or ANCA-associated vasculitis. Aortic stenosis may result in a delayed carotid upstroke, whereas a bounding pulse may indicate aortic insufficiency or a hyperdynamic circulation, as seen in advanced cirrhosis, hyperthyroid states, or left-to-right shunting. An assessment of jugular venous pressure should be made in all patients as a surrogate for right atrial pressure; prominent v waves within the venous pulse contour indicate the presence of tricuspid regurgitation.
Cardiovascular
For obvious reasons, a careful cardiac examination is required in all patients with suspected PH. A prominent pulmonary component of the second heart sound (P2) heard best over the left sternal border in the second to fourth intercostal space is present in most patients with idiopathic PAH, but requires practice to appreciate [13]. Regurgitant murmurs related to tricuspid, and less commonly pulmonic, insufficiency may be found. An RV lift and right-sided S4 are clues to RV hypertrophy. A right-sided S3 indicates compromised RV function and correlates with reduced cardiac output [13]. Right-sided gallops are best appreciated along the left sternal border. Fixed splitting of the second heart sound is observed in the setting of an ASD; a pansystolic murmur may indicate the presence of a VSD, whereas a constant, machine-like murmur is classically associated with a PDA. Murmurs associated with either aortic or mitral valve pathology and S3 or S4 gallops heard best towards the apex suggest underlying left-sided heart disease. Systemic hypotension with a narrow pulse pressure, tachycardia, and cool extremities are ominous signs of a low cardiac output state. Patients suspected of having left-to-right shunting should undergo a search for arteriovenous communications, including iatrogenic fistulae such as those created intentionally for hemodialysis or inadvertently during femoral vascular cannulation.
Pulmonary
Patients with idiopathic, heritable, or toxin-mediated PAH typically have an unremarkable pulmonary examination. In patients with PH due to left-sided heart disease and cardiogenic edema, rales may be appreciated along with dullness to percussion signifying the presence of pleural effusions; the latter finding may also be associated with liver disease. Fine, Velcro-type rales suggest fibrotic lung disease. Wheezing or rhonchi should prompt consideration of chronic obstructive pulmonary disease. Patients with chronic thromboembolic disease may exhibit a pulmonary artery bruit, caused by turbulent blood flow through narrowed proximal pulmonary arteries. The bruit is heard best by auscultation of the interscapular area during a breath hold.
Gastrointestinal
Hepatomegaly, pulsatility of the liver, and ascites may be seen in the setting of chronic elevations in right heart pressure due to PH of any cause. Concomitant splenomegaly or the presence of caput medusae suggests underlying portal hypertension, whereas isolated splenomegaly may be indicative of a lymphoproliferative disorder. Ascites can be a presenting manifestation of constrictive pericarditis. Occasionally, patients with PAH treated with epoprostenol may develop ascites in the absence of portal hypertension [85]. Epoprostenol therapy also has been associated with progressive splenomegaly when used for the treatment of patients with portopulmonary hypertension [86, 87].
Skin, Muscles, and Joints
A thorough musculoskeletal and skin examination should be performed with particular attention to findings that may implicate an underlying connective tissue disease, such as skin thickening, sclerodactyly, rash, telangiectasias (which could also indicate liver cirrhosis), synovitis, or ulcerative lesions. Clubbing of the digits may be seen with interstitial or chronic hypoxemic lung disease, cystic fibrosis, bronchiectasis, thoracic malignancy, congenital cyanotic heart disease, pulmonary veno-occlusive disease (PVOD), and chronic inflammatory states such as inflammatory bowel disease or endocarditis. Muscle wasting may reflect generalized malnutrition or neuromuscular disease. In patients receiving vasodilator therapy for PAH (in particular prostacyclin analogues), flushing of the skin may be noted.
Diagnostic Testing
Echocardiography
In contemporary practice, 2DE remains the most common modality leading to suspicion of PH and the most common screening tool used to assess patients suspected of having PH. It can readily estimate pulmonary arterial systolic pressure (assuming certain conditions are met) and also provides supplementary anatomic and physiologic information that can help elucidate the underlying pathophysiologic mechanisms responsible for PH as well as gauge its severity vis-à-vis right heart function. 2DE is best used as a screening tool when PH is suspected due to the presence of certain symptoms or examination findings. 2DE is discussed in more detail in Chap. 9, but a brief discussion will be presented here.
When faced with an echocardiogram reporting elevations in pulmonary arterial systolic pressure (PASP), the clinician must attempt to answer several questions:
1.
Is the reported PASP accurate?
2.
If accurate, does the patient indeed have PH?
3.
If PH is present, what is the etiology?
4.
To what degree is the presence of PH impacting circulatory function?
2DE is an imperfect diagnostic test with limitations in both sensitivity and specificity. Nevertheless, an understanding of its strengths and limitations will allow the clinician to risk-stratify a patient with cardiopulmonary symptoms and help direct the remainder of the diagnostic evaluation.
Echocardiography laboratories typically calculate and report PASP using the modified Bernoulli principle, according to the following equation:
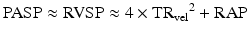 PASP will equal RV systolic pressure (RVSP) in the absence of pulmonic valve stenosis. TRvel and RAP denote Doppler-derived velocity of the tricuspid regurgitation (TR) jet and an estimate of right atrial pressure, respectively. While there is some debate as to the upper limit of normal of 2DE-derived PASP, a value of 40 mmHg is reasonable based on the available evidence, although older subjects may require a higher cutoff [1, 88]. In carefully studied populations, there is a strong correlation between 2DE- and RHC-derived values for PASP using traditional linear regression analysis when comparative measurements are obtained in close temporal proximity [89, 90]. However, suboptimal visualization or improper Doppler interrogation of the TR jet may lead to inaccurate estimates of TRvel; the error associated with this inaccuracy is then magnified exponentially. Also, some patients will have insufficient TR to measure TRvel. Estimates of RAP based on an assessment of inferior cava collapsibility are also prone to error [91]. Contemporary studies of PH referral populations using Bland-Altman analysis suggest that ~50 % of 2DE-derived PASP estimates are inaccurate; both over- and underestimation occur, though the magnitude of underestimation appears to be greater [91, 92]. Thus, 2DE estimates of PASP should be viewed as simply one piece of information as part of a broader 2DE assessment of the right heart–pulmonary circulation–left heart axis.
PASP will equal RV systolic pressure (RVSP) in the absence of pulmonic valve stenosis. TRvel and RAP denote Doppler-derived velocity of the tricuspid regurgitation (TR) jet and an estimate of right atrial pressure, respectively. While there is some debate as to the upper limit of normal of 2DE-derived PASP, a value of 40 mmHg is reasonable based on the available evidence, although older subjects may require a higher cutoff [1, 88]. In carefully studied populations, there is a strong correlation between 2DE- and RHC-derived values for PASP using traditional linear regression analysis when comparative measurements are obtained in close temporal proximity [89, 90]. However, suboptimal visualization or improper Doppler interrogation of the TR jet may lead to inaccurate estimates of TRvel; the error associated with this inaccuracy is then magnified exponentially. Also, some patients will have insufficient TR to measure TRvel. Estimates of RAP based on an assessment of inferior cava collapsibility are also prone to error [91]. Contemporary studies of PH referral populations using Bland-Altman analysis suggest that ~50 % of 2DE-derived PASP estimates are inaccurate; both over- and underestimation occur, though the magnitude of underestimation appears to be greater [91, 92]. Thus, 2DE estimates of PASP should be viewed as simply one piece of information as part of a broader 2DE assessment of the right heart–pulmonary circulation–left heart axis.
Because the hemodynamic definition of PH requires knowledge of the mean PAP, PASP from 2DE cannot be used to define the presence or absence of PH. Although techniques exist to derive mPAP from acceleration time within the RV outflow tract (RVOT) [93], they are not commonly reported on most echocardiograms. Alternatively, mPAP may be estimated using formulas based on PASP [94, 95]; however, such methods are not recommended to replace invasive determination of mPAP by RHC, wherein other hemodynamic variables pertinent to the evaluation of patients with suspected pulmonary vascular disease can be obtained.
If the 2DE-derived PASP is consistent with PH, perhaps the most critical issue the clinician faces is determining the likelihood that the PH is arterial (i.e., PAH) vs. venous (related to left atrial hypertension) in origin, given the ramifications for prognosis and management. In such cases, RHC is required to confirm (or refute) the suspicion and guide therapy. While RHC has a relatively low risk of complications, performing RHC on all patients with elevated PASP on 2DE is neither practical nor desirable. At present, the ideal approach to noninvasively gauge the pretest probability of pulmonary vascular disease so as to optimize use of RHC is unknown. In practice, this decision is influenced by several factors, including the patient’s risk of harboring PAH, results of other noninvasive testing (discussed below), and features on 2DE.
Beyond estimation of PASP, a thorough survey of parameters of right and left heart chamber size and function can be extremely helpful in assessing the likelihood that PAH is present. In general, the greater the extent of abnormalities related to right heart size and/or function, the higher the likelihood of PAH. When abnormalities in left heart size and/or function dominate the echocardiographic picture, there is a greater likelihood of pulmonary venous hypertension [96]. Simple indices of right heart pressure or volume overload easily seen on an apical four-chamber view include an RV:LV size ratio >1, flattening or leftward bowing of the interventricular septum, and encroachment of the RV into the apex. The presence of a mid- or late-systolic “notch” within the RVOT Doppler flow velocity envelope strongly predicts the presence of a PVR >3 Wood units [97]. A reduced tricuspid annular plane systolic excursion (TAPSE) is an easily obtainable surrogate of compromised RV longitudinal shortening and cardiac output, and has prognostic value in PAH [98–100]. When such findings are seen in the absence of significant left heart abnormalities, the clinician should increase his/her suspicion that PAH is present. In contrast, left ventricular systolic or diastolic dysfunction, left ventricular enlargement or hypertrophy, left atrial enlargement, or significant aortic or mitral valvular disease suggests a substrate for increased left atrial pressure and thus pulmonary venous hypertension. A 2DE-based prediction rule incorporating left atrial size, Doppler-based parameters from the RVOT (acceleration time, presence of notching), and lateral mitral E:e′ (a surrogate for left atrial filling pressure) was recently shown to have an area under the receiver operator characteristic (ROC) curve of 0.92 for the identification of PAH [101]. A pericardial effusion, while not specific for PAH, has been consistently associated with a poor prognosis in this group [102–105]. Lastly, 2DE (with or without agitated saline contrast) may disclose evidence of an intracardiac shunt, which could be the cause of PH (e.g., ASD, VSD, PDA), or, in the case of a PFO, may explain significant shunt-type hypoxemia in a patient with idiopathic or other types of PAH and elevated right heart pressures. If suspicion of an intracardiac shunt is high but transthoracic 2DE is nondiagnostic, transesophageal echocardiography [106] or cardiac magnetic resonance [107] should be performed. A comparison of various 2DE parameters which can help the clinician distinguish pulmonary arterial vs. venous hypertension is provided in Table 8.4.
Table 8.4
Echocardiographic parameters to distinguish pulmonary arterial from pulmonary venous hypertension
Pulmonary arterial hypertension | Pulmonary venous hypertension |
|---|---|
2-D Imaging | |
Normal LV ejection fraction | Reduced LV ejection fraction |
Normal or size or small (underfilled) LV | Dilated LV |
Dilated RA/Normal LA | Normal RA/Dilated LA |
No LVH | LVH |
RV:LV size ratio > 1 | RV:LV size ratio < 1 |
RV apex-forming | RV not apex-forming |
Reduced RV function/TAPSE | Normal RV function/TAPSE |
Doppler imaging | |
Grade I or no diastolic dysfunction | Grade II–III diastolic dysfunction |
Minimal or no mitral/aortic valve disease | ≥Moderate mitral/aortic valve disease |
Systolic notching of FVE in RVOT | No systolic notching of FVE in RVOT |
AcT of FVE in RVOT <70 ms | AcT of FVE in RVOT >95 ms |
The issue of diastolic dysfunction in the context of PH deserves additional comment. The clinician should recognize that diastolic dysfunction may worsen with exercise due to further impairment in filling time secondary to increased heart rate, and as such may be underappreciated as a cause of exertional dyspnea based on 2DE parameters obtained at rest. In such situations, assessment of diastolic function with exercise 2DE may be informative [108]. While diastolic dysfunction appears to be very common, particularly in the elderly or those with other comorbidities as discussed previously, patients with PAH due to pulmonary vascular disease may also exhibit impaired left ventricular relaxation as a consequence of pathologic right → left ventricular interaction [66, 109]. A recent study of idiopathic/heritable PAH patients found an 88 % prevalence of Grade 1 diastolic dysfunction (impaired relaxation) [68]. Thus, the simple presence of mild diastolic dysfunction or impaired relaxation in the setting of an elevated PASP should not be considered synonymous with pulmonary venous hypertension. Rather, the clinician should attempt to integrate this finding with the overall clinical picture and echocardiographic assessment of left and right heart function to arrive at an assessment of the likely etiology of PH.
Using 2DE as a screening tool in populations considered at high risk for PAH has been recommended by various professional societies, although this is largely based on expert opinion [6–8]. Regular screening with 2DE seems reasonable for patients with a family history of PAH or with known BMPR2 mutations given that ~20 % of carriers will ultimately develop PAH [110, 111]. In dyspneic patients with scleroderma but without significant lung disease, ~8 % will have PAH by RHC [112]. Screening of patients with the scleroderma spectrum of diseases is recommended. The DETECT study provides useful guidance in this regard [113]. Screening methods in this setting can include a combination of PFTs with diffusion capacity for carbon monoxide (DLco), serum levels of brain natriuretic peptide (BNP) or its N-terminal pro-hormone (NT-proBNP), and echocardiography. As mentioned previously, given the elevated morbidity associated with uncontrolled portopulmonary hypertension in the setting of liver transplantation, 2DE should be performed in all patients being considered for this procedure [37].
Chest X-ray (CXR)
Given its relative low cost and ease, the CXR can provide useful information in the evaluation of patients with suspected PH; however, its suboptimal sensitivity precludes its use as a primary screening modality [114]. General CXR findings of PH include enlarged main pulmonary arteries and right-sided cardiac chamber enlargement; however, such changes may not be apparent in less advanced PH. In the NIH primary PH registry, 90 % of subjects displayed prominence of the main pulmonary artery [13]. An obliterated retrosternal air space as visualized on the lateral projection specifically suggests RV hypertrophy. When PAH is present and related to increased PVR, rapid tapering (or “pruning”) of the pulmonary arteries may be seen. In contrast, when PH is related to increases in left atrial pressure or high pulmonary flow from an anatomic shunt, increased vascular markings and Kerley B lines may be observed; a similar pattern is associated with PVOD where the resistance to blood flow through the small pulmonary veins can result in areas of increased pulmonary capillary pressure. Focal areas of oligemia or an abrupt cutoff of a pulmonary artery should raise suspicion for CTEPH [115], particularly when pleural abnormalities are present which may represent sequelae of prior infarcts [116, 117]. Calcification of the pulmonary arteries has been described in congenital heart disease with reversed shunts (Eisenmenger syndrome) [118], and occasionally large pulmonary artery aneurysms may develop in cases of long-standing PAH. CXR may be particularly useful in assessing for associated conditions that may cause PH, such as hyperinflation (COPD), increased reticulation/cystic changes/honeycombing (interstitial lung disease/pulmonary fibrosis), or reduced lung volumes that may be associated with hypoventilation (obesity hypoventilation syndrome, neuromuscular, or skeletal conditions).
Electrocardiogram (ECG)
Like CXR, the ECG is a readily obtainable ancillary tool that may corroborate a suspicion for PH, but is of limited sensitivity, particularly in the detection of early disease [114]. While a majority of patients with significant PAH will display findings of right axis deviation, RV hypertrophy or RV strain, up to 13 % of patients will have a normal ECG [13, 119]. Recently, a QRS duration of ≥120 ms was found to be an independent predictor of mortality in a small cohort of IPAH subjects [120]. When evaluating a patient with known PH who is clinically deteriorating, the ECG can confirm suspicion of a tachy- or bradyarrhythmia as a contributing factor.
Pulmonary Function Tests (PFTs) and Arterial Blood Gas (ABG) Analysis
PFTs are commonly obtained in the evaluation of the dyspneic patient. In the context of suspected PH, they are extremely important in determining whether airway or parenchymal lung processes are present which may account for PH (i.e., Group 3 PH). A mild restrictive defect may be seen in IPAH [13, 121] and may be related to bronchial and vascular smooth muscle hypertrophy [122] or respiratory muscle weakness [123, 124]. Interestingly, IPAH has also been associated with peripheral airway obstruction [125, 126]. Thus, mild perturbations in pulmonary function indices should not immediately implicate the presence of intrinsic lung disease, but should trigger consideration of such potential etiologies; in such cases, chest CT can be very useful in refining the clinical assessment. The attribution of PH to intrinsic parenchymal or airway disease engenders uncertainty when spirometric or lung volume parameters fall within moderate grades of dysfunction, as studies have not typically proven a strong correlation between the severity of lung disease and the presence or severity of PH, although PH is typically associated with more advanced lung disease [43, 127–133]. Recently, PH has been identified as a significant complication of the combined pulmonary fibrosis and emphysema syndrome [134, 135]. These patients may display relatively preserved (“pseudonormalized”) spirometry and lung volumes due to the counterbalancing effects of coexistent restriction and obstruction, but typically have significant reductions in DLco and abnormal CT imaging.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


