Anticoagulation for Cardiopulmonary Bypass
Linda Shore-Lesserson
Alan Finley
Glenn S. Murphy
Glenn P. Gravlee
HISTORY
Numerous events led to the first performance of surgical procedures with cardiopulmonary bypass (CPB) in Philadelphia in 1953 (1). Development of an effective and reversible method to prevent blood clotting in an extracorporeal circuit impeded this development. Heparin was discovered accidentally in 1916 by an ambitious young medical student named Jay McLean, who had been assigned to experiment with cephalin, a thromboplastic substance. McLean investigated extracts of the heart and liver to determine if the thromboplastic substance found in the brain extracts might be something other than cephalin. Using similar extraction procedures, he discovered an extract that retarded plasma coagulation from both the heart (named cuorin) and the liver (named heparphosphatide initially, then changed to heparin) (2,3). Although McLean had made an important scientific discovery at a young age, he subsequently selected a clinically oriented career that apparently precluded his participation in the development of heparin as a drug (4).
The purification of heparin proceeded in the 1920s, and a fairly crude preparation was first used to anticoagulate blood for transfusion in 1924. Febrile reactions curbed this application, and it took another 12 years to attain a heparin preparation that appeared safe for intravenous administration. During that interval, the discovery that heparin could be obtained from bovine lung less expensively than from bovine liver proved practical in the commercial development of heparin. Clinical trials in thrombotic disorders were initiated in 1935, and it was evident even then that heparin could prevent clot formation or extension, although it possessed minimal ability to dissolve existing clots. Chargaff and Olson (5) discovered in 1937 that the peptide protamine dramatically neutralizes the anticoagulant effects of heparin. Gibbon (6) reported heparin-induced anticoagulation for CPB in animals in 1939. These events led to the selection of heparin for anticoagulation and protamine for its subsequent neutralization in the first human operation in which CPB was used, in 1953. Although most other aspects of CPB practice have changed markedly since that time, the use of heparin and protamine has continued for over 60 years. This longevity serves as a testimonial to the astute judgment of Gibbon and his colleagues at Jefferson Medical College. Consequently, this chapter primarily discusses the pharmacology and clinical use of heparin for this purpose. Strategies for monitoring the anticoagulant effects of heparin and of heparin alternatives are also presented. The final portion of the chapter is devoted to discussion of heparin-induced thrombocytopenia (HIT) and heparin-induced thrombocytopenia with thrombosis (HITT) syndromes.
HEPARIN PHARMACOLOGY
Structural Characteristics and Biologic Function
Heparin, more specifically described as a glycosaminoglycan, is a polysaccharide that resides almost exclusively in mast cells. Its physiologic purpose, however, remains uncertain, although roles in neovascularization and in the inflammatory response appear likely. Clearly, endogenous heparin plays no significant role in maintaining the fluidity of circulating blood. Heparan, a related glycosaminoglycan having a substantially lower sulfur content, dangles tantalizingly from endothelial cell membranes to attract circulating antithrombin III (ATIII) irresistibly and potentiate thrombin inhibition. Physiologic anticoagulation at the blood-tissue interface thereby derives not from heparin but from heparan. The primary physiologic purpose of heparin may be to participate in nonimmunologic defense against bacterial infections, with other roles likely in capillary angiogenesis and lipid metabolism.
Most heparin preparations can be described as unfractionated, which means that the heparin compound isolated from animal tissues contains heparin molecules of various lengths, with molecular weights ranging from 3,000 to more than 40,000 Da. The mean molecular mass approximates 15,000 Da (7,8) (Fig. 19.1). The molecular weight distribution varies somewhat with the tissue source, animal source, and method of purification (9,10). This variability has some clinical relevance because the spectrum of clinical actions of heparin derives in part from the molecular weight distribution of a heparin compound (11). As a result, each unfractionated commercial heparin preparation might best be described as a family of drugs with actions and potency that may vary from batch to batch (12,13,14).
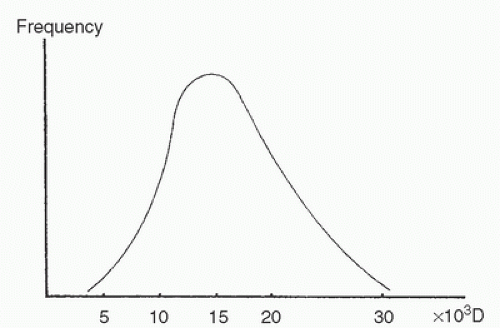 FIGURE 19.1. Molecular weight distribution pattern of a typical commercial preparation of unfractionated heparin. (From Stiekema JCJ. Heparin and its biocompatibility. Clin Nephrol 1986;26(Suppl 1):S3-S8, with permission.) |
Heparin can be distinguished from other polysaccharides by its acid nature. It is the strongest macromolecular acid in the body, a characteristic that derives from abundant sulfation of its saccharide units. The basic subunit consists of a repeating disaccharide that contains a uronic acid residue linked to a glucosamine residue (Fig. 19.2). Both the uronic acid and glucosamine residues can assume many different forms based on the side groups attached to these hexose units. Sulfate groups may attach to the hexose ring through an oxygen, amino, aminoacetyl, or methane link. The result is a large molecule that bears a highly negative charge within the physiologic pH range, and that therefore attracts positively charged molecules. Specific saccharide sequences along the heparin chain determine binding sites to other macromolecules for which it has high affinity, such as ATIII, thrombin, and lipoprotein lipase. In the case of ATIII, a specific pentasaccharide sequence binds consistently to a specific amino acid sequence on the ATIII molecule (15).
Tissue Source and Commercial Preparation
Heparin can be purified from several tissues and from several mammalian species. Although the name heparin was selected because the substance was originally isolated from liver extracts, intestinal mucosa and lung tissue represent the most common commercial sources. Mucosal heparin tends to have a lower mean molecular weight, a more cross-linked polysaccharide structure, and a lower cost than lung heparin (12). Differences in the tissue source from which heparin is extracted influence molecular structure and activity more than differences in the animal source (16,17). However, nuclear magnetic resonance spectroscopy has enabled the identification of differences between porcine and bovine mucosal heparin in sulfation of various subunits. Nuclear magnetic resonance spectroscopy has also demonstrated a higher content of 3-O-sulfated glucosamine units (active site for ATIII binding) in porcine than in bovine mucosal heparin (18). Heparin manufacturers most often extracted mucosal heparin from pigs (porcine mucosal heparin) and lung heparin from cattle (bovine lung heparin).
Both porcine mucosal heparin and bovine lung heparin had been widely used as anticoagulants for CPB. Both provide effective anticoagulation and prevent thrombosis in experimental models (19) and in adult volunteers (20). Investigating standardized needle punctures in exposed carotid arteries in dogs, Abbott et al. (21) found a higher incidence of delayed hemorrhage in animals anticoagulated with mucosal heparin than with lung heparin. This difference remained significant even when the anticoagulation was neutralized with protamine. Two studies prospectively compared mucosal and lung heparin for CPB in humans. Stewart and Gaich (22) found that higher doses of mucosal heparin than of lung heparin were needed to reach the desired prolongation of the activated clotting time (ACT), a finding that might be attributable to batch variability. In a dialysis study comparing mucosal with lung heparin at standard patient doses of heparin, a lower anticoagulant activity was noted in International Units/mL with bovine heparin, despite higher weight-based drug levels. Both preparations resulted in anticoagulant effects and plasma levels within the recommended safe range (23). Fiser et al. (24) observed greater postoperative blood loss in patients randomly assigned to receive mucosal heparin than in those who received lung heparin. Both animal and human studies indicate that mucosal heparin can be neutralized with 25% to 30% less protamine than can lung heparin, a consequence of the lesser anti-IIa activity of mucosal heparin than bovine (17,25). These studies used standard tests of plasma coagulation such as clotting time and activated partial thromboplastin time (aPTT), which might not detect residual unneutralized inhibition of factor Xa. Because of its lower mean molecular weight, mucosal heparin, in concert with ATIII, more effectively inhibits factor Xa than does lung heparin (26), and protamine only partially neutralizes this effect. The higher mean molecular weight of lung heparin enables this molecule to inhibit factor IIa more effectively through ATIII. The anti-IIa activity is more susceptible to protamine antagonism, which may explain the greater propensity for delayed bleeding found after anticoagulation with mucosal heparin (20,22) and the requirement for lower doses of protamine for neutralization. Fairly limited prospective comparisons and some theoretic considerations therefore suggest a slight advantage of lung heparin for CPB anticoagulation, but many hospitals have ceased to utilize lung heparin because of its greater propensity for HIT. In the mid-1990s, bovine heparin production and commercialization ceased in the United States and many European countries due to fears of potential transmission of bovine spongiform encephalopathy. Porcine heparin became the exclusive heparin product used in medicine, hemodialysis, and cardiac surgery. Porcine heparin is primarily derived from Chinese production sources, and the reliance on a single source of drug is very limiting and increases the risk of drug shortage. This became evident in 2008
when the heparin produced in China was found to be contaminated with oversulfated chondroitin sulfate, and was responsible for catastrophic hypotensive events in cardiac surgery and hemodialysis patients. A serious heparin shortage ensued as there were no other manufacturers making heparin in the quantities being produced by China. In addition, current detection methods for transmissible BSE agents are superior now compared with those during the mid-1990s (when mad cow disease reached crisis numbers), and some believe that a reintroduction of bovine heparin into the US commercial market is in order. Both clinical studies and practice demonstrate that for CPB anticoagulation, clearly both mucosal and lung heparin are effective and safe.
when the heparin produced in China was found to be contaminated with oversulfated chondroitin sulfate, and was responsible for catastrophic hypotensive events in cardiac surgery and hemodialysis patients. A serious heparin shortage ensued as there were no other manufacturers making heparin in the quantities being produced by China. In addition, current detection methods for transmissible BSE agents are superior now compared with those during the mid-1990s (when mad cow disease reached crisis numbers), and some believe that a reintroduction of bovine heparin into the US commercial market is in order. Both clinical studies and practice demonstrate that for CPB anticoagulation, clearly both mucosal and lung heparin are effective and safe.
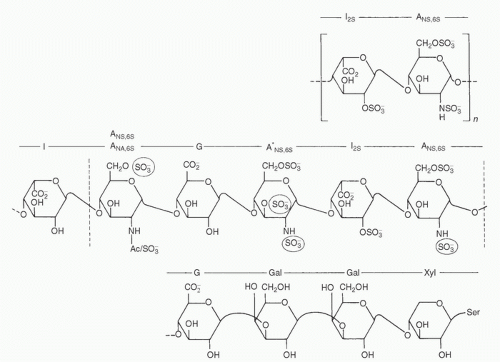 FIGURE 19.2. Molecular structure of different parts of the heparin polysaccharide chain. The top row demonstrates a common repeating disaccharide subunit consisting of L-iduronic acid 2-sulfate (I2S) and N-sulfo-α-D-glucosamine 6-sulfate (ANS,6S), which represents up to 90% of beef lung heparin and 70% of porcine mucosal heparin. In the middle row, the five saccharide units between the vertical dotted lines comprise the pentasaccharide sequence required for binding antithrombin III (ANA,6S substitutes an N-acetyl for the N-sulfo group on ANS,6S, G represents β-D-glucuronic acid, and I represents β-L-iduronic acid). This sequence occurs in approximately 33% of the chains of mucosal heparin and approximately 20% of the chains of lung heparin. The circled sulfate groups are believed essential for high-affinity binding. The bottom row shows the typical terminal sequence of a heparin molecule (Gal, galactose; Xyl, xylose) linking to a serine amino acid residue. (From Casu B. Methods of structural analysis. In: Lane DA, Lindahl U, eds. Heparin. Chemical and biological properties, clinical applications. Boca Raton, FL: CRC Press, 1989:25-49, with permission.) |
Because of the acid nature of heparin, a ligand must be bound to it when the compound is prepared for commercial use. Sodium and calcium have been used for this purpose. The two salts are indistinguishable with intravenous heparin administration, but the calcium salt retards the uptake of subcutaneously administered heparin (9,27) and may reduce local hematoma formation with subcutaneously administered heparin.
Potency Standardization
Four assays have been used in recent years to determine the potency of unfractionated heparin (UH) (19,28), including International, United States, British, and European standards. The International Standard represents the mean of the pharmacopoeial methods, which results in some variation in potency between international units (IU) and United States Pharmacopoeia (USP) units. The USP assay defines 1 USP unit as the amount of heparin that maintains the fluidity of 1 mL of citrated sheep plasma for 1 hour after recalcification. The
British Pharmacopoeia (BP) method uses sulfated ox blood activated with thromboplastin. The BP method has been superseded by a European Pharmacopoeia (EP) method, which recalcifies sheep plasma in the presence of kaolin and cephalin incubated for 2 minutes, thereby constituting an aPTT for sheep plasma. The EP method rigorously standardizes the collection of sheep plasma, which might diminish the assay variability previously reported between batches of sheep plasma substrate (16). Although speculation exists about the clinical significance of batch-to-batch variability in heparin potency, it seems more likely that pharmacodynamic differences account for most of the observed variations in the clinical anticoagulation response to heparin (see subsequent section on Heparin Resistance). Because the relation between mass (milligrams) and potency (units) varies among heparin preparations, it appears more sensible to record heparin doses in units than in milligrams.
British Pharmacopoeia (BP) method uses sulfated ox blood activated with thromboplastin. The BP method has been superseded by a European Pharmacopoeia (EP) method, which recalcifies sheep plasma in the presence of kaolin and cephalin incubated for 2 minutes, thereby constituting an aPTT for sheep plasma. The EP method rigorously standardizes the collection of sheep plasma, which might diminish the assay variability previously reported between batches of sheep plasma substrate (16). Although speculation exists about the clinical significance of batch-to-batch variability in heparin potency, it seems more likely that pharmacodynamic differences account for most of the observed variations in the clinical anticoagulation response to heparin (see subsequent section on Heparin Resistance). Because the relation between mass (milligrams) and potency (units) varies among heparin preparations, it appears more sensible to record heparin doses in units than in milligrams.
Pharmacokinetics
Because heparin administration for CPB is exclusively intravenous, this discussion is limited to that route of administration. After central venous injection of a heparin bolus, the onset of maximal ACT prolongation in the radial artery occurs within 1 minute. A previous study (29) suggested that heparin action peaks 10 to 20 minutes after administration in cardiac surgical patients, but this finding probably was an artifact representing prolongation of the ACT by other factors, such as hemodilution and hypothermia. Controlling for these factors, another study clearly demonstrated that the onset of action of heparin is much faster, and maximal ACT prolongation probably occurs in less than 5 minutes (30). A rapid redistribution effect probably accounts for a modest reduction in the anticoagulant effect of heparin that occurs 3 to 13 minutes after the peak effect (Fig. 19.3). It remains possible that the onset of action would be slightly delayed in states of low cardiac output or with peripheral venous injection.
Distribution
Heparin is macromolecular and highly polarized, so one would expect minimal distribution beyond the bloodstream. These principles and the results of bioassay studies of heparin kinetics were the basis for the former belief that the distribution of heparin is virtually confined to the plasma compartment of the bloodstream (31,32). Substantial in vitro evidence now points to the redistribution of heparin into the endothelial cells (33,34,35,36), although this redistribution appears small in comparison with that of most other drugs. Uptake into extracellular fluid, alveolar macrophages, splenic and hepatic reticuloendothelial cells, and vascular smooth muscle also occurs (37,38,39). Some or all of these tissues create a relatively small reservoir for heparin that probably contributes to the delayed recurrence of heparin-induced anticoagulation (heparin rebound) after protamine neutralization of the heparin residing in the bloodstream. Defining an apparent volume of distribution for heparin remains elusive because pharmacokinetic studies in humans have used some measure of heparin effect (i.e., a bioassay) rather than direct measurement of plasma heparin concentration. Bioassays represent the most practical approach to the clinical evaluation of heparin pharmacodynamics, but these assays can only indirectly and crudely assess pharmacokinetics.
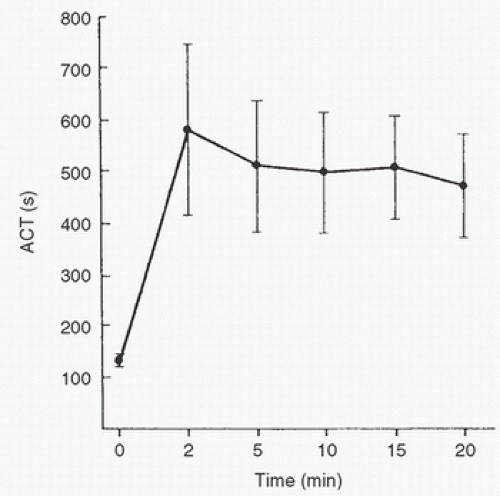 FIGURE 19.3. The activated clotting time (ACT) measured through radial artery blood sampling at five different intervals after the injection of 300 units of heparin per kilogram into the right atrium. Maximum ACT prolongation occurred within 2 minutes in most patients, with subsequent moderate ACT reduction, likely reflecting a rapid redistribution effect. (From Gravlee GP, Angert KC, Tucker WY, et al. Early anticoagulation peak and rapid distribution after intravenous heparin. Anesthesiology 1988;68:126-129, with permission.) |
Elimination and Excretion
Heparin elimination has been studied by using various clotting times as bioassays, so the available information largely defines the time course of its clinical activity. The clotting times differ in their sensitivities to heparin, so differences reported in heparin “elimination” kinetics under similar conditions are not necessarily inconsistent. Despite these unavoidable limitations, several aspects of heparin pharmacokinetics can be explained by existing studies. Very small doses of heparin produce minimal or no effect, suggesting a rapid initial clearance, possibly the result of the affinity of heparin for endothelial membranes (40). The doses used for CPB anticoagulation are very large, and the biologic elimination half-life of heparin is dose-dependent (41,42). In the only volunteer study in which doses sufficient for CPB were used, Olsson et al. (41) found a half-life of 126 ± 24 minutes with a heparin dose of 400 units/kg. The half-lives for doses onefourth and one-half that large were 61 ± 9 and 93 ± 6 minutes, respectively. Because CPB distorts the bioassay methods
through hypothermia and hemodilution, attempts to define heparin pharmacokinetics in patients undergoing CPB are largely impractical. Hypothermia delays heparin elimination, with virtually constant heparin concentrations shown during 40 to 100 minutes of CPB at 25°C (43). Wright et al. (44) found a progressive decline in heparin concentrations at all temperatures, but the rate of decline was delayed in proportion to the hypothermia. Bull et al. (45) found a small decrease in the rate of ACT decline at 30°C (in comparison with normothermia), although the independent prolongation of ACT by hypothermia had not been discovered at the time of that study. Mabry et al. (46) found the consumption of heparin to vary from 0.01 to 3.86 units/kg/min during CPB. In addition, there is little correlation between the initial ACTbased sensitivity to heparin and the rate of heparin decay (47). In children, the variability in ACT response is greater than that in adults and is highly dependent on the test mechanism and activators used (48) (Fig. 19.4).
through hypothermia and hemodilution, attempts to define heparin pharmacokinetics in patients undergoing CPB are largely impractical. Hypothermia delays heparin elimination, with virtually constant heparin concentrations shown during 40 to 100 minutes of CPB at 25°C (43). Wright et al. (44) found a progressive decline in heparin concentrations at all temperatures, but the rate of decline was delayed in proportion to the hypothermia. Bull et al. (45) found a small decrease in the rate of ACT decline at 30°C (in comparison with normothermia), although the independent prolongation of ACT by hypothermia had not been discovered at the time of that study. Mabry et al. (46) found the consumption of heparin to vary from 0.01 to 3.86 units/kg/min during CPB. In addition, there is little correlation between the initial ACTbased sensitivity to heparin and the rate of heparin decay (47). In children, the variability in ACT response is greater than that in adults and is highly dependent on the test mechanism and activators used (48) (Fig. 19.4).
Severe renal impairment may also prolong heparin action, although available studies yield conflicting results. Liver disease apparently has little effect on heparin elimination. The primary mechanism for heparin elimination remains uncertain, but metabolism in the reticuloendothelial system and renal elimination both occur (37,39,40).
Pharmacodynamics
Inhibition of Fibrin Formation
Heparin induces anticoagulation primarily by potentiating the activity of ATIII (or “antithrombin”), a plasma glycoprotein (GP) with a molecular weight of 58,000 Da. Heparin attaches to a lysine residue on ATIII, thereby altering the ATIII configuration and rendering it much more attractive to thrombin (15,49). Heparin therefore increases the thrombin-inhibitory potency of circulating ATIII by a factor of 1,000 or more. In addition to converting fibrinogen to fibrin enzymatically, thrombin activates cofactors VIII and V, thereby greatly increasing the rate of fibrin clot formation through the intrinsic and common pathways, respectively. Figure 19.5 shows the plasma coagulation cascade, which can be somewhat crudely divided into intrinsic, extrinsic, and common pathways. ATIII also inhibits factors IXa, Xa, XIa, and XIIa (50) kallikrein, and plasmin. Plasma coagulation factors vary in their sensitivity to ATIII and to different chain lengths of heparin. UH inhibits thrombin most quickly, then inactivates Xa, IXa, XIa, and XIIa with progressively decreasing rate constants (50). Thrombin inhibition involves transient simultaneous heparin binding to ATIII and to thrombin, which requires a relatively long oligosaccharide chain. This is why shorter heparin chains are relatively ineffective in thrombin inhibition, even if they contain the pentasaccharide sequence required for ATIII binding (Fig. 19.6). Factor Xa inhibition involves heparin binding to ATIII, which in turn binds to factor Xa molecules that need not bind separately to heparin, as depicted in Figure 19.6. Two-thirds or more of the heparin molecules present in commercially available preparations have no anticoagulant effect (11), which probably results from an absence of the specific pentasaccharide sequence that binds to ATIII.
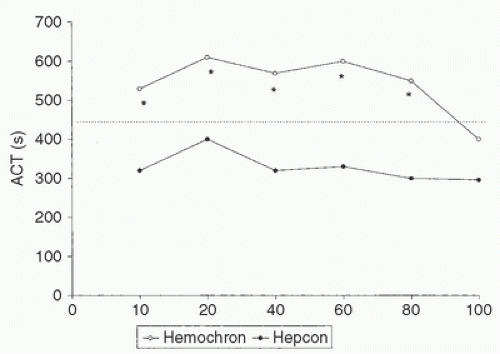 FIGURE 19.4. The Hemochron and HemoTec (Hepcon) activated clotting time (ACT) values in 22 pediatric patients at six time-points during cardiopulmonary bypass (CPB). Hemochron ACT was significantly higher than HemoTec ACT at five time-points, and HemoTec ACT was below the threshold value of 400 seconds at all time-points. *p < 0.01. (Modified from Horkay F, Martin P, Rajah M, et al. Response to heparinization in adults and children undergoing cardiac operations. Ann Thorac Surg 1992;53:822-826, with permission.) |
Heparin also binds to cofactor II, a GP of 65,000 Da that inactivates thrombin independently of ATIII (51). This reaction occurs more slowly and requires higher heparin concentrations than does thrombin inhibition through the heparin-ATIII complex. The thrombin-heparin-cofactor II interaction is significantly catalyzed in vitro by plasma heparin concentrations between 0.1 and 0.4 units/mL, which is well below the heparin concentrations used for CPB. This mechanism might therefore contribute routinely to the anticoagulant effect of heparin during CPB, and might assume particular importance in patients with ATIII deficiency.
Variability of Patient Response
Whether heparin concentration or a clotting time is measured, the response to a fixed-dose heparin bolus varies substantially from patient to patient (29,45,52,53,54,55,56,57,58,59). The variability of clotting time responses usually exceeds the variability of blood- or plasma heparin concentrations observed, as demonstrated by Monkhouse et al. (52) as early as 1953 (53,54,60). Bull et al. (45,61) showed a 3-fold variation in ACT response to a heparin bolus of 200 units/kg, and Esposito et al. (53) found a 4-fold ACT range (62-267 seconds) at that dose and a 6-fold range (128-755 seconds) after an intravenous bolus of 400 units/kg. Bull et al. (45,61) reported equally impressive ranges of heparin elimination rates during CPB when using ACT as an indirect heparin assay.
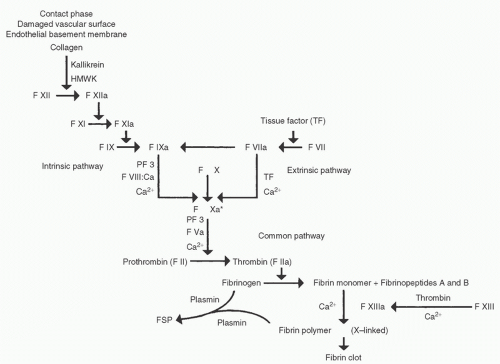 FIGURE 19.5. Schematic diagram of the plasma coagulation pathway, divided into intrinsic, extrinsic, and common component pathways. F, factor; roman numerals, different coagulation factors by number; a, activated coagulation factor; Ca2+, ionized calcium; HMWK, high-molecular-weight kininogen; PF3, platelet factor 3; FSP, fibrin split products. |
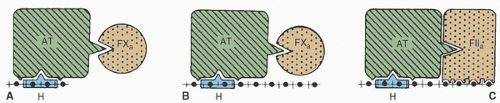 FIGURE 19.6. Schematic representation of the interaction between heparin (H) and activated factors X (FXa) and II (thrombin, FIIa). Inhibition of factor IIa requires a heparin chain containing at least 18 saccharide units (shown in B and C) in addition to the critical pentasaccharide sequence for antithrombin III (ATIII) binding (shown as the framed portion of H bound to AT). This occurs because the heparin molecule must simultaneously bind ATIII and thrombin. Factor Xa inhibition is also accomplished with polysaccharide chains long enough to inhibit factor IIa (shown in B), but shorter chains can also inhibit factor Xa (shown in A with an octasaccharide chain) because simultaneous binding of heparin, ATIII, and factor Xa is not required for Xa inhibition. (From Holmer E. Low-molecular-weight heparin. In: Lane DA, Lindahl U, eds. Heparin. Chemical and biological properties, clinical applications. Boca Raton, FL: CRC Press, 1989:575-595, with permission). |
Side Effects
Plasma coagulation, the formation of a platelet plug, and fibrinolysis constitute the three major components of blood clot formation and dissolution. The therapeutic effects of heparin derive primarily from its effects on plasma coagulation, described in the preceding text, but heparin also affects the other two components.
Heparin-induced activation of fibrinolysis has been identified in a primate model (62). During simulated CPB in a
baboon model, heparin administration led to activation of fibrinolysis as measured by plasmin activity, immunoreactive plasmin light chain, and immunoreactive fibrinogen fragment E (63). The mechanism whereby heparin facilitates fibrinolysis may involve the direct stimulation of plasminogen activator release from endothelial cells and monocytes. Additional modes of activation include indirect stimulation of plasminogen activator by release of protein C, inhibition of fibrin polymerization (64), modulation of antiplasmin effects, and enhancement of the effects of tissue factor pathway inhibitor (TFPI). Activation of the fibrinolytic pathway does occur during anticoagulation associated with CPB, so heparin might participate in this activation.
baboon model, heparin administration led to activation of fibrinolysis as measured by plasmin activity, immunoreactive plasmin light chain, and immunoreactive fibrinogen fragment E (63). The mechanism whereby heparin facilitates fibrinolysis may involve the direct stimulation of plasminogen activator release from endothelial cells and monocytes. Additional modes of activation include indirect stimulation of plasminogen activator by release of protein C, inhibition of fibrin polymerization (64), modulation of antiplasmin effects, and enhancement of the effects of tissue factor pathway inhibitor (TFPI). Activation of the fibrinolytic pathway does occur during anticoagulation associated with CPB, so heparin might participate in this activation.
Heparin also has numerous effects on platelets, many of which occur acutely, and these would therefore be relevant in patients undergoing CPB. Table 19.1 lists the aggregatory effects of heparin on platelets, which most often represent laboratory findings obtained in human platelet-rich plasma. The clinical significance of these findings is uncertain, but clearly heparin binds avidly to platelets. With the use of specific chemical and enzymatic treatments to produce heparin-derived glycosaminoglycans, the platelet-binding properties of heparin have been elucidated (77). Although specific receptors for heparin on the platelet surface have not been identified, heparin binding relates directly to molecular weight and sulfation and is unrelated to ATIII affinity (77,78,79,80,81). Heparin binding decreases with the decreasing size of the heparin fragment and is therefore clinically negligible in the low-molecular-weight heparin (LMWH) preparations. Small quantities of heparin may bind to the GPIIb/IIIa platelet receptor, but this is not the major locus for heparin binding. At clinical doses, heparin induces a platelet release reaction characterized by release of platelet factor 4 (PF4), GPIIb/IIIa activation, P-selectin expression, and increased aggregation that is not seen with LMWH derivatives (75,82). At plasma levels as high as 100 units/mL, heparin suppresses degranulation and P-selectin expression, a finding that indicates the future possibility of separation of the anticoagulant and the antiplatelet properties of heparin (76,83). Moderate prolongation of bleeding time and transient decreases in platelet count have been present in some investigations and absent in others (80). Both predictable and idiosyncratic effects of heparin on platelets have been reviewed by Warkentin and Kelton (80). The idiosyncratic syndrome of HIT is discussed later in a separate section (see Heparin-Induced Thrombocytopenia).
TABLE 19.1. Acute effects of heparin on platelets | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In clinical settings other than CPB, bleeding complications comprise the most common side effect of heparin, with reported incidences varying from 1% to 37% (84,85,86,87). The profound anticoagulation present during CPB probably increases surgical bleeding, but blood salvage through the cardiotomy suction usually renders this unimportant. Large doses of heparin induce fibrinolysis and activate platelet activation to contribute to abnormalities of hemostasis (88). Conversely, insufficient heparin anticoagulation during CPB causes consumption of coagulation factors, which may also result in a bleeding diathesis (89). Excessive postoperative bleeding from residual unneutralized heparin or from heparin rebound can occur after CPB and should be closely monitored (see Chapter 20).
The intravenous heparin bolus dose administered before CPB decreases arterial pressure and systemic vascular resistance approximately 10% to 20% without affecting cardiac output or heart rate (90,91). Urban et al. (91) related this change to a decrease in ionized calcium levels, and they were able to prevent them by prophylactically administering 125 mg of calcium chloride.
Heparin can induce a variety of metabolic and immunologic effects (92) that might contribute to the array of abnormalities known to occur in those systems during CPB. Heparin dramatically increases plasma levels of lipoprotein lipase by releasing this enzyme from vascular endothelium. This activity bears no relation to ATIII affinity, and it results in triglyceride degradation and increased circulating levels of free fatty acids. Although the clinical significance of these effects remains unclear, they could potentially affect myocardial metabolism and the plasma-free fraction of lipid-soluble drugs.
Rare acute reactions attributed to heparin include anaphylaxis, pulmonary edema (93,94,95,96,97,98,99), and disseminated intravascular coagulation (100). Some of these reactions have been traced to the preservatives chlorocresol and chlorbutol (95,96). Harada et al. (93) reported the uneventful use of bovine lung heparin for CPB in a child who had previously experienced anaphylaxis from porcine mucosal heparin, and
Schey (101) reported resolution of a localized skin reaction to subcutaneous heparin injections when LMWH was substituted for UH. Interactions with antibiotics have also been reported, notably with gentamicin and erythromycin (102,103), although these appear unlikely to have clinical significance. Administration of heparin for weeks to months has been associated with osteoporosis, alopecia, hyperaldosteronism, and benign elevation of serum glutamic oxaloacetic transaminase levels (104,105,106).
Schey (101) reported resolution of a localized skin reaction to subcutaneous heparin injections when LMWH was substituted for UH. Interactions with antibiotics have also been reported, notably with gentamicin and erythromycin (102,103), although these appear unlikely to have clinical significance. Administration of heparin for weeks to months has been associated with osteoporosis, alopecia, hyperaldosteronism, and benign elevation of serum glutamic oxaloacetic transaminase levels (104,105,106).
HEPARIN DOSING
During the first two decades of cardiac surgery with CPB, heparin dosing was accomplished empirically, with initial doses usually ranging from 200 to 400 units/kg and maintenance doses of 50 to 100 units/kg given as often as every 30 minutes or as infrequently as every 2 hours (51). The priming fluid used for the extracorporeal circuit usually contained 10,000 to 20,000 units of heparin. Heparin monitoring, defined as laboratory testing for the adequacy of blood heparin concentration or of a heparin-induced anticoagulant effect, was limited by the absence of an easily applicable test. This situation changed with the introduction of two coagulation tests that could be performed practically at the bedside on whole blood. The activated coagulation time (more appropriately called the activated clotting time [ACT] in later publications) was introduced by Hattersley (107), and the blood-activated recalcification time (BART), also termed the recalcified whole-blood partial thromboplastin time, was introduced by Blakely (108). Both of these tests were first reported for CPB heparin monitoring in 1974, BART for cardiac surgery and ACT for longterm respiratory support (56,109). The two classic papers by Bull et al. (45,61) pointed out the apparent inadequacy of empiric heparin and protamine dosing protocols and recommended a structured approach using the ACT. This served as a turning point for heparin management during CPB, and the application of ACT monitoring to CPB evolved from virtually nonexistent to widespread during the ensuing 5 years. Laboratory tests for heparin monitoring are covered in Chapter 18.
Clinical Assessments and Comparisons of Heparin Dosing and Monitoring Techniques



