Fig. 10.1
Pulmonary arterial hypertension time-course and type of vascular remodeling pattern secondary to exposure to the monocrotraline pyrrole. A single subcutaneous injection of monocrotaline at a dose of 60 mg/kg causes non-reversible PAH associated with vascular remodeling mainly characterized by media muscularization. Black arrow shows time of injection. X marks time of tissue harvesting. Chemical structure of monocrotaline was obtained from Wikimedia Commons (Public domain) (Micrograph from lung vessel was modified with permission from Gomez-Arroyo et al. [3])
The MCT model offers great advantages against other models of PAH such as cost, accessibility, and reproducibility. The response to MCT is very dose sensitive, such that an injection of 40 mg/kg of MCT will yield modest PAH, whereas a single dose of 60 mg/kg will produce more severe PAH at 3 weeks, and significant mortality after 4–5 weeks [15]. Importantly, MCT induces PAH without the need for other exposures such as hypoxia, which requires the housing of animals in a nitrogen dilution chamber that might not readily available and, in turn, can be costly to acquire and maintain. In addition, because chronic hypoxia is not required, the model can be readily characterized in a longitudinal fashion, allowing researchers to study the impact of mild to severely increased pulmonary afterload upon the development of right ventricular hypertrophy by a variety of invasive and non-invasive means [16, 17] as well as the impact upon exercise adaptation [18], for example. Similar to tissues obtained from human PAH, tissues from MCT-induced PAH exhibit exaggerated TGF-β signaling and pro-fibrotic transcriptional activity, and suppressed expression of BMPR2 and downstream BMP signaling activity [19]. Based on this molecular signature, as well as histology, the MCT model has been suggested to be particularly helpful for highlighting the contributions of fibrosis, inflammation, and lung injury in pulmonary vascular disease [3], a notion that is supported by the observation that numerous experimental and approved therapies with potential anti-fibrotic and anti-inflammatory effects have demonstrated efficacy in the MCT model [19–24]. Because MCT pro-toxin requires metabolism by the liver the generate an active metabolite, and because MCT can also cause hepatic inflammation and hepatotoxicity, experimental design in this model must account for the potential effects of MCT on the metabolism and pharmacokinetics of experimental drugs, and conversely, exogenous agents or experimental conditions which alter the function of hepatic cytochrome P450 monooxygenases may modulate the availability and pulmonary vascular toxicity of MCTP [13]. MCT may exert direct toxicity on the heart or other organs by impacting the microcirculation of those organs, with potentially confounding effects that make it difficult to isolate the effects of pressure-overload on the right ventricle versus direct toxic effects on the right ventricle, for example. The activity of MCT is generally believed to be a species-specific phenomenon in the rat. In earlier studies, administration to mice of as much as 600 mg/kg of MCT, tenfold greater than those required to elicit pulmonary vascular remodeling in the rat, was observed to generate pneumotoxicity without significant pulmonary vascular remodeling or right ventricular hypertrophy [25], a finding attributed to the lack of cytochrome P450 monooxygenases in mice required to convert MCT to MCTP. However, in more recent studies, similarly high doses of MCT, or direct administration of MCTP have been reported to generate successful models of PAH in standard mouse backgrounds, as well as genetically modified mice [26–28]. While the use of MCT/MCTP in mice would provide significant new opportunities for investigation, further investigation may be needed to standardize MCT or MCTP regimens and their consequent phenotypes in this species.
SUGEN 5416-based Models
SUGEN 5416 (SU5416), 1,3-Dihydro-3-[(3,5-dimethyl-1H-pyrrol-2-yl)methylene]-2H-indol-2-one also known as semaxinib, (Fig. 10.2) is a potent ATP-competitive inhibitor of VEGF receptor kinases 1 and 2 [29], which was originally developed for use as an adjuvant anti-angiogenic agent in the treatment of cancer by the former SUGEN company in the late 1990s [30]. At the same time, it was observed that VEGF and VEGFR2 mRNA and protein are overexpressed in the obstructive and plexiform lesions of human and rodent pulmonary arterial hypertension, the histology of which were suggested to resemble a process of disordered angiogenesis [31–33]. SU5416 was initially administered to rats to test the hypothesis that VEGF is required for the maintenance of airways, and was found to induce alveolar septal cell apoptosis, emphysema, as well as pruning of the distal pulmonary vasculature [34]. When the same group tried to combine the effect of VEGF receptor blockade with chronic hypoxia in order to worsen emphysema, they observed, surprisingly, that rats exposed to SU5416-hypoxia developed severe vascular remodeling (Fig. 10.2) and non-reversible PAH associated with severe right ventricular dysfunction, right ventricular failure and death when left untreated [35]. In contrast to hypoxia alone and MCT-induced models, this model resulted in neointimal and plexiform lesions populated by proliferating endothelial cells, and was thus one of the first animal models to recapitulate these pathognomonic features of human pulmonary vascular disease [36]. The fact that chronic hypoxia in combination with an anti-angiogenic VEGFR2 inhibitor would cause PAH might seem counterintuitive, given the angiogenic activity and overexpression of VEGF signaling molecules observed at sites of remodeling [37]. Since treatment with a pan-caspase inhibitor prevented the development of angio-obliterative pulmonary hypertension after SU5416-hypoxia exposure [35], a mechanism invoking endothelial cell apoptosis as a triggering event was postulated [38]. Subsequently, in-vitro studies have bolstered the concept that proliferation of apoptosis-resistant endothelial cells contributes to PAH in the SU5416-hypoxia model [39], providing additional evidence for the “cancer paradigm” as a model for understanding the pathophysiology of PAH as a proliferative disorder [38, 40].
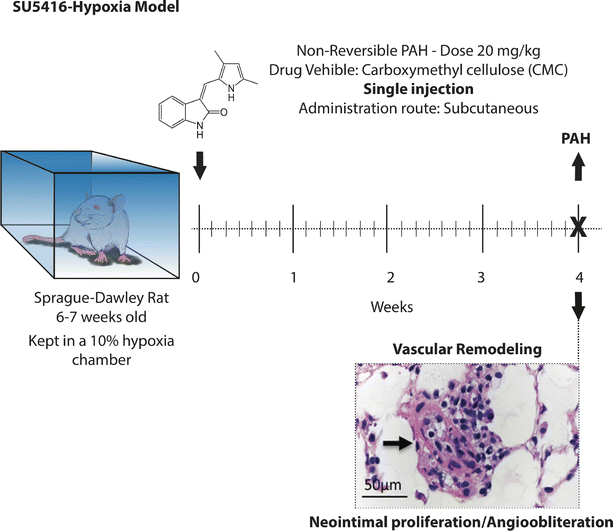
Fig. 10.2
Pulmonary arterial hypertension time-course and type of vascular secondary to exposure to SU5416/Hypoxia. A single subcutaneous injection of SU5416 at a dose of 20 mg/kg in combination with chronic hypoxia (10 %) exposure causes severe, irreversible PAH at 4 weeks, characterized by angioobliteration and muscularization. Black arrow shows time of injection. X marks time of tissue harvesting. Chemical structure of SU5416 was obtained from Wikimedia Commons (Public domain) (Micrograph from lung vessel was modified with permission from Gomez-Arroyo et al. [3])
The SU5416-hypoxia model has permitted the introduction of contemporary scientific concepts to understanding further the factors regulating PAH pathobiology, such as the contributions of pulmonary endothelial cell mitochondrial fragmentation [41], endothelial apelin-fibroblast growth factor signaling dysregulation [42], and dysregulation of multiple miRNA species [43]. This model has helped to elucidate the requirement for copper [44] and thyroid (T4) hormone [45] for the development of angioproliferative lesions. Recently, SU5416-hypoxia was used to demonstrate the role of Forkhead box O (FoxO) transcription factor 1 in the regulation of cell cycle control and bone morphogenic protein receptor type 2 (BMPR2) signaling in experimental PAH [46].
Two key advantages that the SU5416-hypoxia model has provided to the field of PAH-research are that (i) this model does not require genetic manipulation, underscoring the critical role of VEGF in adult lung biology, and (ii) that severe pulmonary vascular remodeling occur even in the absence of chronic hypoxia [47]. Not too long after the first report of SU5416-hypoxia-induced PAH was published, attempts to generate alternative models of PAH prompted researchers to combine SU5416 with other insults or “hits” [47]. Several of these modifications have resulted in seminal contributions to field, in particular, the athymic-SU5416 rat model of PAH. Taraseviciene-Stewart, Nicolls, Voelkel and collaborators demonstrated that administration of a single dose of SU5416 to athymic nude rats lacking T cells resulted in severe angio-obliterative PAH and RV dysfunction even in the absence of chronic hypoxia [48]. This model yielded an early opportunity to evaluate the role of chronic inflammation and immune dysregulation in a histologically faithful model of PAH, providing evidence for the role of regulatory T cells [49] and macrophage-derived proinflammatory mediators [50] in the pathogenesis of PAH [51].
The SU5416-hypoxia model has also been studied extensively to evaluate the pathobiology of chronic right ventricular (RV) dysfunction secondary to PAH. The RV in the SU5416-hypoxia rat model responds to pulmonary hypertension with robust hypertrophy, followed by dysfunction and systolic failure [52]. Based on data derived from this model, it has been proposed that maladaptive/dysfunctional RV hypertrophy is characterized by capillary rarefaction [53], myocardial ischemia [54], cardiomyocyte apoptosis and fibrosis [55, 56], and significant metabolic remodeling and mitochondrial dysfunction [57]. These molecular and cellular changes are associated with decreased cardiac output, marked RV dilatation and decreased exercise capacity. Importantly, the administration of SU5416 by itself does not induce capillary rarefaction in the right ventricle, despite its potential effects on the myocardial microcirculation, whereas pure pressure overload by pulmonary arterial banding elicits pathophysiologic effects similar to that seen with SU5416-hypoxia, supporting the relevance of the SU5416-hypoxia model in studying RV pathophysiology.
Like any other experimental model of disease, there are several limitations that should be considered when utilizing SU5416-hypoxia for study in PAH. For example, neither chronic hypoxia nor VEGF blockade has been implicated directly in human PAH. Based on the dysregulation of VEGF signaling activity in affected tissues, however, a potential role of imbalance between angiogenic and anti-angiogenic activity has been posited [37]. In contrast to the effects of SU5416, administration of imatinib, a small molecule multi-kinase inhibitor targeting an overlapping set of tyrosine kinase growth factor receptors including VEGFR and PDGFR, has been proposed as a therapy for limiting the progression of PAH [58]. Multi-kinase inhibitors such as nilotinib and dasatinib with overlapping effects on PDGFR and Src were found to ameliorate experimental PAH more potently than imatinib [59]. Yet nilotinib has been associated with peripheral arterial occlusive disease in man [60, 61], and more vexingly, dasatinib has been associated with incident PAH in previously unaffected patients receiving these drugs for cancer therapy [62]. The relationship of VEGF/VEGFR signaling to the many other signaling axes targeted by these small molecules, and their pathogenetic roles remains incompletely defined. While SU5416 has clearly highlighted the importance of VEGF and VEGFR signaling as a possible trigger for PAH, it is not known if environmental factors might engender similar effects upon this signaling axis to contribute to the development of PAH in man. Other smaller, but non-trivial considerations accompany the use of this animal model are cost and equipment. Requiring rats, inhibitor compound, and access to chronic hypoxia facilities, the cost of generating the SU5416-hypoxia model is significantly greater than that of monocrotaline. Moreover, akin to MCT, there appears to be some species specificity for SU5416-hypoxia, which requires the use of rats for induction of the PAH histopathophenotype. Some researchers have reported a similar phenotype when the SU5416-hypoxia model is employed in mice [63], affording the opportunity to explore a potentially robust PAH model in genetically modified mice. However, several factors should be considered when using SU5416-hypoxia in mice: (1) the degree of vascular remodeling is not the same as that of rats, lacking neointmal remodeling [64], (2) the disease in mice is reversible upon return to normoxia [63] and (3) severe RV failure has not been documented [4].
Strain-Specific and Genetically-Modified Models of PAH
The Fawn-hooded rat (FHR) strain has been known to be exquisitely sensitive to the development of PAH and pulmonary vascular remodeling in response to mild or moderate hypoxia in comparison to other rat strains [65, 66], and develops spontaneous PAH at 20 weeks of age, culminating in mortality at 60 weeks of age [67]. The sensitivity of FHR to hypoxia has been attributed to mitochondrial abnormalities, particularly in the pulmonary artery smooth muscle, preceding the development of PAH and persisting in culture. A relative deficiency in mitochondrial reactive oxygen species production appears to cause aberrant activation of hypoxia inducible factor (HIF-1α), which in turn inhibits the expression of oxygen sensing, voltage-gated potassium channels such as Kv1.5, leading to a disruption of normal oxygen sensing to mimic chronic hypoxia [67]. A number of oxygen-sensitive candidate genes potentially affected by the known rearrangement of chromosome 1 in FHR have been suggested to contribute the molecular mechanism of the PAH phenotype. This model has been particularly useful in highlighting potential contributions of mitochondrial and metabolic function to the pathogenesis of PAH, as well as therapeutic interventions acting upon these pathways.
A recent transgenic mouse model has tested a longstanding and controversial hypothesis that inflammation and immune dysregulation contribute to the pathogenesis of PAH [68]. Patients with idiopathic or familial PAH have been observed to express increased circulating levels of IL-1β, IL-6, IL-8, IL-10, and TNF-α, with levels of IL-6, IL-8, and IL-10 being associated with increased mortality [69, 70]. The fact that IL-6 is also elevated in other inflammatory diseases associated with PAH including systemic lupus erythematosus suggested a particularly important role of this cytokine [71]. Steiner and colleagues showed that a transgenic mouse expressing IL-6 under the control of the Clara cell promoter in the lung did not develop pulmonary hypertension spontaneously, but developed severe PAH (with an average RVSP of 60 mmHg) and right ventricular hypertrophy following exposure to chronic hypoxia [72]. In fact the vascular remodeling seen in this context, which included neointimal remodeling, had some features that were reminiscent of human plexiform lesions, including overexpression of VEGF, as well as of pro-survival proteins such as survivin and Bcl-2. It is not known if these mice will spontaneously regress upon return to normoxia, or under other experimental therapies. Given the potential relevance to human PAH associated with immune dysregulation, whether or not this model might be responsive to therapeutic modulation of inflammation is an important subject of further investigation.
Heterozygous loss-of-function mutations in BMPR2, expressing the bone morphogenetic protein type II receptor (BMPRII) are known to occur in 75–80 % of patients with familial or heritable forms of PAH, 10–25 % of patients with sporadic idiopathic PAH [73–78], with a reduced penetrance of approximately 20 %. Testing whether or not this mutation might be sufficient to engender PAH in animal models has been a long-term pursuit in the field. West and colleagues showed in 2004 that overexpression of a dominant negative BMPRII transgene in the smooth muscle of mice led to increased muscularization of pulmonary arterioles, and increased right ventricular systolic pressures at Denver altitude [79]. While expression at high levels of a dominant negative BMPRII transgene suppressed BMP signaling and led to pulmonary vascular disease, it was acknowledged that this model might not necessarily recapitulate the functionally haploinsufficient state found in most mutation-positive humans [76, 77]. In 2005, Beppu and colleagues found that a Bmpr2 heterozygous knock-out mouse developed minimally increased right ventricular systolic pressures under normoxia or hypoxia, and with very modest changes in alveolar capillary structure [80]. These haploinsufficient mice developed more significant pulmonary hypertension and medial hypertrophy of distal vessels following intratracheal gene transfer of 5-lipoxygenase, suggesting that inflammation might unmask a latent phenotype in these animals. Since BMPR2-null mice are arrested in early development [81], conditional gene targeting of Bmpr2 in mice [82] combined with endothelial-specific recombination via an endothelial (Alk1 promoter driven) Cre recombinase was required to test the effects of postnatal, tissue specific ablation [83]. These endothelial targeted conditional Bmpr2-null mice were reported to have spontaneous PAH, albeit with variable penetrance, accompanied by increased distal pulmonary arteriolar muscularization and mild right ventricular hypertrophy. This phenotype of endothelial targeted conditional Bmpr2-null mice, despite being sporadic and generally mild, has been revisited in the context of therapy by other groups. Attenuation of leukocyte recruitment and extravasation by the inhibition of chemokine receptors CXCR1 and 2 was sufficient to normalize pulmonary hypertension in this model [84]. In a study by Spiekerkoetter and colleagues, pulmonary hypertension was exacerbated in this model by chronic hypoxia, and the resulting PAH and right ventricular hypertrophy was normalized by administration of FK506, based on a mechanism of enhanced BMP signaling via the inhibition of BMP type I receptor regulatory molecule FKBP12 [85]. The studies using endothelial ablation of Bmpr2 in conditional knockout mice importantly demonstrated that complete ablation of BMPRII from the vascular endothelium was by itself insufficient to generate consistently severe PAH or remodeling by itself, yet could be additive with other insults. These studies leave open the role of BMPRII loss-of-function in other vascular tissues (e.g., smooth muscle) or tissue compartments, and other potential environmental insults that might contribute.
Systemic-to-Pulmonary Shunting, Pneumonectomy, Overflow and Banding Models
It has been long been recognized that left-to-right sided shunts, such as those associated with congenital heart disease, or systemic veno-venous shunts, such as those associated with portopulmonary syndrome, can be associated with PAH. Eisenmenger syndrome, a condition that commonly results from unrepaired post-tricuspid congenital cardiac defects – such as ventriculoseptal defects or patent ductus arteriosus – has challenged many researchers to evaluate the role of flow-induced shear stress in the development of vascular remodeling [38].
Models of over-circulation-induced PAH have been described in the rat by means of an aorto-caval shunt [86], or alternatively, by a single injection of monocrotaline after the removal of a lung lobe (pneumonectomy) by White and colleagues [87]. This combination results not only in the development of pulmonary hypertension but also the development of neontimal proliferative lesions, a feature not seen with MCT treatment alone.
Because of size restrictions, most of the animal models of flow-induced pulmonary hypertension have been performed in medium to large size animals such as miniature swine, piglets and domesticated pigs. Rondelet, Naeije and collaborators demonstrated that anastomosis of the left subclavian artery to the pulmonary arterial trunk in piglets resulted in pulmonary arterial hypertension and lung vascular remodeling characterized mainly by vessel muscularization [88]. Interestingly, this model exhibits other features associated with human PAH such as increased levels of circulating vascular endothelial growth factor and plasma endothelin-1 level, decreased expression of BMPRII and endothelin-1 receptors [89] and has even been associated with right ventricular failure [90]. The development of right ventricular failure in this model contrasts with observations from Eisenmenger syndrome cohort studies, in which signs or symptoms of heart failure are not observed until late in disease. The piglet shunt model develops severe RV dysfunction in the setting of relatively mild pulmonary hypertension, leading some to speculate whether or not RV dysfunction might be a more direct consequence of decreased expression of angiogenic factors angiopoietin-2 and VEGF, and the increased expression of pro-inflammatory cytokines interleukin (IL)-1α, IL-1β, and tumour necrosis factor-α (TNF-α) that are observed in the RV tissues of these animals [89, 90].
Recently a model of PAH was described using Yorkshire pigs subjected to repeated distal pulmonary arterial embolization with dextran beads and selective coiling of proximal pulmonary arteries with silk suture material [91]. This procedure elicited acute and sustained increases in pulmonary arterial pressures, with the development of pulmonary vascular remodeling, accompanied by decreased right ventricular function and right ventricular hypertrophy. In order to model postcapillary pulmonary hypertension, pulmonary vein banding has also been applied in the pig model, leading to robust increases in pulmonary arterial pressures and pulmonary vascular resistance, accompanied by distal pulmonary arterial remodeling, and RV dysfunction [92]. While pulmonary hypertension was a consistent finding in this model, a subsequent analysis of the natural history of pigs subjected to pulmonary vein banding revealed subsets which developed right ventricular failure, and those which remain compensated [93], suggesting that this model could be used to help understand factors which dictate the variable natural history of PH in man.
Thus, swine-based models of PH have been advantageous particularly for the study of shunt-based, overflow-mediated, or obstructive forms of pulmonary vascular disease. The high cost of maintaining these larger animals, and the paucity of commercially available reagents for performing genetic and protein analysis on porcine tissues are major limitations, however. A key advantage of swine based models, however, is the ability to perform survival surgery, critical for studying mechanical interventions such as shunt closure or right ventricular assist devices. Larger animals such as miniature and domesticated swine, by virtue of the human-like proportions of their cardiopulmonary system, permit sophisticated and detailed invasive measurements of hemodynamics, and imaging techniques for assessing myocardial function by approaches similar if not identical to those deployed in human disease [91–93].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


