Fig. 19.1
Relationship between different strains of rats whose selective crossing over several generations gave rise to different animal models of diabetes, hypertension, obesity and metabolic syndrome. Currently male obese ZDF, obese ZSF1, Koletsky and obese SHHF/Mcc-fa cp rats are accepted as good animal models of metabolic syndrome. *: the timing depends on the gender and genotype. CHF congestive heart failure, DCM, dilated cardiomyopathy, DD diastolic dysfunction, DM diabetes mellitus, HF heart failure, HGly hyperglycemia, HLep hyperleptinemia, HLip hyperlipidemia, HIns Hyperinsulinemia, HT hypertension, IR insulin resistance, MI myocardial infarction, mth months, RF renal failure, SD systolic dysfunction, wks weeks
Of note is the fact that most of diet-induced ATH models do not present occlusive coronary artery disease, myocardial infarction, cardiac dysfunction and premature death, which are a rather common feature of human atherosclerotic disease that may lead to congestive HF.
19.2.2 Metabolic Syndrome
The metabolic syndrome (MS) is diagnosed whenever three of the following disorders: obesity, hypertension, insulin resistance, hyperglycemia or dyslipidemia are found in the same individual. This triad represents a threatening and complex disorder, which is the leading cause of CV morbidity and mortality in modern societies. Over the last four decades, two significant mutations (i.e. fa and cp) that elicit obesity have been identified in rats. The fa mutation, originally described by Zucker in 1961 [4] has been backcrossed into several strains (Fig. 19.1). The cp mutation described originally by Koletsky has been also backcrossed into several strains, including SHHF/Mcc-fa cp rats [5]. Several strains have been selectively inbred and backcrossed for many generations to select particular features, such as hypertension, obesity or hyperglycemia, giving rise to a huge variety of models whose relationship can be difficult to establish and comprising a part or the whole metabolic syndrome phenotype as illustrated in Fig. 19.1.
This chapter will address separately animal models of hyperglycemia and diabetes mellitus, hypertension and obesity, highlighting those that might represent good animal models of the metabolic syndrome.
19.2.2.1 Hyperglycemia and Diabetes Mellitus
Animal models have been extensively used in diabetes research, including pharmacological rodent models of diabetes such as streptozotocin (STZ) and alloxan, drugs that when administred to rats or mice induce diabetes mellitus (DM) as soon as 24–48 h post-injection. These substances are selectively toxic to pancreas β-cells, induce insulin deficiency and hyperglycemia and therefore represents a model of type-1 diabetes [6]. On the other hand, selective inbreeding has produced several strains of animal that are considered reasonable models of type 2 diabetes and related phenotypes such as obesity and insulin resistance (Fig. 19.1 and Table 19.1). Apart from their use in studying the pathogenesis of the disease and its complications, all new treatments for diabetes, including islet cell transplantation and preventive strategies, are initially investigated in animal models. In recent years, a large number of new genetic animal models for the study of diabetes, including knockin or generalized and tissue-specific knockout mice have been described. Rodent models of type-2 diabetes include the Zucker fatty rat, as well as db/db and ob/ob mice, all of which display dysfunctional or absent leptin homeostasis and therefore develop insulin resistance at different timepoints (Table 19.1).
These rodent models share many features of human diabetic cardiomyopathy. For example, rodent models of obesity, insulin resistance and type 2 diabetes present left ventricle (LV) hypertrophy, diastolic dysfunction, increased cardiac fatty acid uptake and utilization, decreased cardiac efficiency, impaired mitochondrial energetics, increased myocardial lipid storage and impaired Ca2+ handling [7]. Moreover, depending on the model, in vivo studies have revealed susceptibility to systolic and diastolic dysfunction assessed by echocardiography and hemodynamics [8] as well as propensity to ischemia/reperfusion injury following ligation/unligation of the left anterior descending coronary artery (LAD), which occurs in conjunction with structural and functional changes to the LV [9].
However, there are several limitations when comparing these models to human diabetic cardiomyopathy, as spontaneous ischemia and atherosclerotic disease are not prominent in rodents [10]. The latter becomes simultaneously a valuable aspect as the effects of obesity, insulin resistance and diabetes on the heart can be studied independently of coronary artery disease [11]. Importantly, rodent models present sudden and uncontrolled hyperglycemia or insulin resistance, whilst in the clinical setting patients with diabetes are increasingly well controlled. Moreover, because DM develops at varying stages in these models, it is important to keep in mind that studies performed in animals before the onset of diabetes may reflect changes that are secondary to the underlying obesity and insulin resistance, and studies performed after the onset of diabetes may reflect the added effects of hyperglycemia of different durations.
19.2.2.2 Systemic Hypertension
Spontaneously Hypertensive Rats SHR
Systemic hypertension is another relevant risk factor for human congestive HF. Spontaneous hypertension is a natural model of pressure overload, in which systemic hypertension leads to HF with ageing. Hypertensive vascular lesions appear within 6–7 weeks, being more severe in males than in females. For the first 12 months, hypertrophy is compensated and contractility is preserved, but after 18–24 months there is overt congestive HF characterized by fibrosis, LV dilation and reduced systolic function [12]. These structural and functional changes occur together with a marked rise in cytokine levels such as TNF-α and interleukin-6 [13]. Transition to failure has been suggested to depend on significant alterations in the expression of genes encoding extracellular matrix proteins, oxidative stress and increased apoptosis of myocytes [12]. The gradual onset of hypertension with ageing makes this model suitable for studying the transition from hypertrophy to CHF and for reproducing hypertension-induced CHF in humans [14]. It has the advantage of avoiding the complications associated with surgical or pharmacological interventions, while mimicking the changes found in human essential hypertension [12]. Indeed, the SHR is a very useful model of genetic hypertension because drugs that lower blood pressure in SHR also decrease blood pressure in essential hypertension, being this one of the main reasons why researchers have used it extensively. Nonetheless, the long period required for developing CHF poses a great limitation, making it a time-consuming and consequently an expensive model. Moreover, Wistar Kyoto (WKY) is currently accepted as the “normotensive” control of SHR but it shows considerable genetic differences displayed and thus no ideal control exists for SHR.
Table 19.1
Summary of the characteristics of the most usual diabetes mellitus animal models
Model | Type | DM type | Specie | Pathophysiologic changes | Advantages | Disadvantages |
|---|---|---|---|---|---|---|
Alloxan | P | 1 | Mouse | Significant hyperglycemia | Fast, economic and consistent | High mortality rate |
Rat | Ketosis and/or ketoacidosis | Alloxan has a very short half-life (<1 min) | ||||
Glycosuria, hyperlipidemia, polyphagia, polydipsia | Hyperglycemia frequently reverts by pancreatic regeneration | |||||
Neuropathy and cardiomyopathy | ||||||
Streptozotocin | P | 1 | Mouse | Significant hyperglycemia | Fast, economic and consistent | High mortality rate |
Rat | Polyuria, polydipsia | Very severe model | ||||
Muscular atrophy | ||||||
Neuropathy | ||||||
OLETF – Otsuka Long-Evans Tokushima Fatty | G | 2 | Rat | Hyperglycemia | Progressive DM: | Late DM development |
Polyuria, polydipsia | Initial phase (0–9 wk): Pancreatic islet hyperplasia and lymphocytes’ infiltration | Only males develop DM | ||||
Mild obesity | Intermediate phase (10–40 wk): Pancreatic islet fibrosis | |||||
Diastolic dysfunction | Advanced phase (>40 wk): pancreatic islet atrophy. Type 1 DM | |||||
Diabetic nephropathy with nodular glomerulosclerosis (30 wk) | ||||||
ZDF – Zucker Diabetic Fatty | G | 2 | Rat | Dysfunctional leptin receptor | Very common model | Mild nephropathy with hydronephrosis |
Hyperphagia, hyperlipidemia, hyperglycemia | ZDF were selectively bred from Zucker rat which are similar but without hyperglycemia and DM | Due to different genetic backgrounds, no comparisons can be made between ZDF and Zucker rats | ||||
Glucose intolerance and hyperinsulinemia (10–12 wk).that progresses to impaired insulin secretion with β-cells fatigue | ||||||
Mild hypertension and obesity | Females develop DM upon a high-fat diet | Useful for studies on the efficacy of drugs directed at macrovascular function, nephropathy, myocardial injury and lipidemia | ||||
Loss of both GLUT-2 glucose transporters in pancreatic β-cells and GLUT-4 in the muscle (25–55 %) | ||||||
Impaired cardiac contractility and diastolic function (~20 wk) | ||||||
Increased LV mass and fatty acid oxidation | ||||||
Microvascular damage leading to glomerular sclerosis and retinopathy | ||||||
Evidence of vascular dysfunction in the aorta, coronary arteries, and mesenteric arteries in middle-aged obese ZDF rats probably related to frank diabetes | ||||||
GK – Goto-Kakizaki | G | 1 | Rat | Mild fast hyperglycemia | Stable degree of glucose intolerance | Non obese |
Hyperinsulinemia and insulin resistance | Useful for studying advanced diabetic nephropathy | |||||
Decreased insulin production | Wistar rats are the control group | |||||
Retinopathy, microangiopathy, neuropathy and nephropathy | ||||||
Wistar Fatty | G | 1/2 | Rat | Hyperglycemia, hyperlipidemia e hyperinsulinemia (12 wk) | Wistar rats are the control group | Only males develop type-2 DM |
Glucose intolerance and insulin resistance | ||||||
Hypertension (16 wk) | ||||||
Wistar × Zucker selective breeding | Not commercially available | |||||
Db/db | G | 2 | Mouse | Leptin receptor deficiency | Advantages associated with mice reduced size (see text) | Glucose levels progressively increase until the 16th week |
Hyperlipidemia and obesity (8 wk) | ||||||
Polyphagia, polyuria and polydipsia | ||||||
Insulin resistance, hyperinsulinemia (2 wk) and hyperglycemia (8 wk) | ||||||
Peripheral neuropathy | ||||||
Diabetic cardiomyopathy | ||||||
Impaired diastolic function, mitochondrial energetic, Ca2+ homeostasis and cardiac efficiency | ||||||
Impaired endothelial and vascular function | ||||||
Increased LV mass, fatty acid oxidation and RAAS activation | ||||||
Ob/ob | G | 2 | Mouse | Leptin deficiency | Advantages associated with mice reduced size (see text) | Certain degree of infertility |
Hyperphagia and obesity (4 wk) | Allows the evaluation of the early effects of obesity and insulin resistance on cardiac function and the effects of additional hyperglycemia at older ages | Reduced metabolism and hypothermia | ||||
Hyperglycemia and hyperinsulinemia (15 wk, following obesity) | ||||||
Insulin resistance and glucose intolerance | ||||||
Impaired diastolic function, mitochondrial energetic, Ca2+ homeostasis and cardiac efficiency | ||||||
Increased LV mass, fatty acid oxidation and lipid content | ||||||
Pancreatectomy | S | 1 | All | Mild hyperglycemia | Useful for pancreatic regeneration studies | Risk of infection. |
No weight or insulin levels reduction | Can be used in all animal model species | Post-operative precautions | ||||
Avoids pharmacologic toxicity of DM-induction drugs | Digestive complications | |||||
Similar to type-2 DM due to pancreatic degeneration | ||||||
High fat diet C57BL/6 J | HFD | 2 | Mouse | Leptin and insulin resistance | Advantages associated with mice reduced size (see text) | Reduced metabolism and hypothermia |
Hyperphagy and obesity | Present many genetic and environmental features of the human disease | Long high fat diet period | ||||
Hyperglycemia and hyperinsulinemia (following obesity) | Mild hyperglycemia | |||||
Glucose intolerance | High fat diet C57BL/6 J mice change myocardial substrate utilization prior to obesity and severe insulin resistance | |||||
Cardiac dysfunction (20 wk) | Useful for pharmacologic tests | |||||
CIRKO, Akita and OVE 26 | T | 2,1,1 | Mouse | Please check Table 19.2 | ||
The SHR stroke prone (SHR-SP) was selectively bred from a substrain of the SHR that showed even higher levels of blood pressure and a strong tendency to die from stroke [16]. SHR-SP rats also display nephropathy, insulin resistance, hyperinsulinemia, hypertriglyceridemia and hypercholesterolemia. Moreover, despite comparable degrees of hypertension among males and females, stroke-related mortality is greater in males than females. Renal vascular disease including thrombotic microangiopathy affecting glomeruli and microvessels as well as cardiac damage are more prominent in male SHR-SP [17].
Spontaneously Hypertensive HF Prone Rats (SHHF)
These animals were developed by backcrossing the obese SHR (SHROB) rat to the SHR/N rat and afterwards selectively bred to decrease the age at which animals developed CHF. The obese offspring carries the fa cp corpulent gene, which encodes a defective leptin receptor gene, and therefore they develop obesity and HF. The time for the development of HF depends on fa cp genotype and gender (male animals are more prone to HF than females [18]) but, in general, SHHF rats present HF earlier than the SHR strain, with loss of cardiac function starting at the age of 15 months [19]. The obese SHHF display hypertension, nephropathy, obesity, insulin resistance, hyperinsulinemia, type 2 DM, hypertriglyceridemia and hypercholesterolemia. Controls are SHHF lean that also exhibits HF characteristics but at a different age from the SHHF obese. All SHHF, regardless of genotype or gender, eventually develop spontaneous dilated cardiomyopathy (DCM). These animals present alterations in the renin-angiotensin-aldosterone system (RAAS) and also in calcium metabolism [20]. The greatest advantage of this strain, which is considered a good model of metabolic syndrome, is the possibility of studying drug interventions in an extended range of cardiovascular risk factors like obesity, diabetes and renal dysfunction [21].
Dahl Salt-Sensitive Rats
This is a mutant strain of Sprague–Dawley rats that is characterized by hypersensitivity to sodium intake [22]. When placed on a high salt diet since the 6th week of age they develop concentric LV hypertrophy (HYR) without chamber dilation around the 11th week and decompensate HF with marked ventricular dilation between the 15th and the 20th week [23]. Failure is associated with reduced myocardial performance as evidenced by the lower performance of muscle strip preparations and the short lifetime of failing rats [24].
Interestingly, it has been shown that introducing high-salt diet at 7 or 8 weeks of age can result in distinct HF phenotypes. Indeed, the 7-week starting rats showed a steeper elevation in blood pressure and progressive LV hypertrophy, falling into overt HFpEF at approximately 19 weeks. On the other hand, the 8-week starting rats showed a gradual rise in blood pressure and less progressive LV hypertrophy, developing HFrEF at approximately 26 weeks. Therefore, these two different models of overt HF may be useful as models of isolated HFpEF and HFrEF based on the same hypertensive heart disease, which could be relevant to the pathophysiologic and molecular characterization of each HF subtype [25]. Another report found that the development of HF was dissociated from changes in passive diastolic and active systolic properties, suggesting that volume overload plays an important pathophysiological role in development of congestion despite preserved overall ventricular pump function in this model of chronic hypertension [23].
This model is suitable to study the transition from compensated HYR to failure. Moreover, it is often used to identify the role of several pathways and molecular mechanisms like oxidative stress, extracellular matrix degradation, calcium handling impairment, redox regulated transcription factors and apoptotic factors activation [15].
Deoxycorticosterone Acetate (DOCA) Hypertensive Rats
The deoxycorticosterone acetate (DOCA) salt-induced model of hypertension is a typical representative of pharmacologically-induced hypertension. A very high subcutaneous dose of DOCA is required to induce hypertension in rats. Isotonic saline is the sole drinking fluid, which hastens and aggravates the progression to hypertension [26]. Despite being salt-dependent in its initiation, this model frequently needs surgical renal mass reduction or unilateral nephrectomy. DOCA-salt hypertension is a low renin and volume overload form of hypertension. The combination of DOCA-salt and unilateral nephrectomy results in hypertension, renal HYP, nephrosclerosis, cardiac hypertrophy and perivascular fibrosis within 4–5 weeks of chronic treatment [27]. Several studies report diastolic dysfunction in these rats [28].
The pathophysiologic mechanisms underlying the development and maintenance of DOCA-salt hypertension include increased levels of arginine, vasopressin [29], angiotensin II/aldosterone [30], endothelin [31] and oxidative stress [32], excessive activation of the sympathetic nervous system [33] and nitric oxide synthase (NOS) uncoupling due to oxidative depletion of its cofactor tetrahydrobiopterin (BH4) [28]. Nonetheless, the cardiac consequences are minimal during the development of DOCA-salt hypertension-induced HYP [34]. This is in contrast with the decreased cardiac performance reported in other rat models of cardiac HYP and in the failing human heart. Therefore, hypertrophy in hearts of DOCA-salt hypertensive rats does not reproduce the changes observed in the failing human heart [34]. Besides this disadvantage, other DOCA-salt model limitations are (1) the need to employ a large amount of drug, (2) the requirement for surgical reduction of renal mass and (3) the dependence on a strictly controlled ingestion of a high NaCl dose. On the other hand, its advantage is the potential to investigate the role of sodium in the developmental stages of hypertension. In an attempt to overcome some of these limitations, more recently, a group published a mouse model that combines transverse aortic constriction (TAC) with DOCA administration under a normal-salt diet. Compared with TAC mice, TAC plus DOCA mice had similarly normal LV systolic pressure and fractional shortening but more hypertrophy, fibrosis, oxidative stress and diastolic dysfunction with increased lung weights, consistent with HFpEF. Therefore, these authors suggest that pressure-overload HYP sensitizes the heart to mineralocorticoid excess, promoting the transition to HFpEF without activation of the classic mineralocorticoid receptor-dependent gene transcription [35].
Two-Kidneys, One-Clip (2K1C) or One-Kidney, One-Clip (1K1C) Rats
Since 1934, when Goldblatt and his co-workers induced an elevation of blood pressure by partial constriction of the renal artery of the dog [36], many successful models of renal-induced experimental hypertension have been developed in rats. In general, the procedure includes two-kidney Goldblatt hypertension (constriction of one renal artery while the contralateral kidney is left intact) and one-kidney Goldblatt hypertension (one renal artery is constricted and the contralateral kidney is removed) [26]. Clipping one renal artery, while leaving the contralateral kidney untouched, induces systemic hypertension and LV concentric remodeling within 8 weeks [37]. Histologic studies revealed extensive LV fibrosis while echocardiography and hemodynamic data consistently showed diastolic dysfunction [37]. Indeed, inhibition of matrix metalloproteinase activity in these hypertrophic hearts has been shown to provide beneficial effects in terms of structure and function [38]. At cellular level, several changes in the energy metabolism, actin-myosin cross-bridge cycle and protein expression were identified in renovascular hypertensive rats [37]. In addition, the central role of activation of the RAAS [39] and the sympathetic nervous system [40] has been thoroughly studied. In the one-kidney model, no compensatory increase in sodium and water excretion can occur, and hence, fluid volume is retained, which means this model is thus a sodium-fluid volume-dependent model. Therefore, it would be valuable for studying the role of volume expansion in the development of hypertension [41]. During the early developmental stage, if the clip is removed arterial blood pressure returns to normal in both models, suggesting that renovascular hypertension is both reversible and reproducible [42].
ZSF1 Obese Rats
The obese ZSF1 is commonly used to study renal failure with additional disease complications. However, these rats also represent a robust animal model of metabolic syndrome since they comprise hypertension, obesity, type 2 DM, insulin resistance, hyperinsulinemia, hypertriglyceridemia, hypercholesterolemia and even HF. Considering such features, this cardiometabolic risk animal model has recently been shown to develop diastolic dysfunction and HF, having therefore been considered a robust animal model of HFpEF by their 20th week of age. At this age no renal failure was yet observed in obese ZSF1 despite the diastolic dysfunction as confirmed by increased myocardial stiffness mostly due to myofilamentary changes, since myocardial fibrosis was absent. Lean ZSF1 also display hypertension but no diastolic dysfunction, obesity, DM nor the associated metabolic disturbances [43]. Other reports have suggested that obese ZSF1 present decreased LV contractility indices by the 45th week of age.
19.2.2.3 Obesity
Obesity is associated with premature death by increasing the risk of many chronic diseases, including type 2 DM, CV disease and certain cancers. Thus, not surprisingly, many obesity animal models are also diabetic and held major CV abnormalities (Fig. 19.2). As previously mentioned, many available models of obesity derive from selective crossings between rats comprising one out of the two most significant mutations in leptin receptor (i.e. fa and cp). Such is the case of Zucker fatty rats, the fa (fatty) gene “family”, whose obesity is not as marked as in SHROB or JCR:La-cp, the cp (corpulent) gene “family”. Figure 19.1 display several rat models of obesity derived from selective crossings among and between these two “families”. Many obese animal models, such as Zucker fatty rat, SHROB or even SHHF, are also models of hypertension or MS and thus were mentioned in previous sections. Another example is the Otsuka Long-Evans Tokushima Fatty (OLETF) rat obtained by selecting spontaneously diabetic rats exhibiting polyuria, polydipsia and obesity from an out-bred colony of Long-Evans rats. These rats exhibit obesity, hyperglycemia (at the adult age, 18 weeks), hyperinsulinemia, chronic DM, which is first non-insulin dependent but that over time progresses to insulin dependent. The control strain, LETO appears to share some of diabetogenic genes with OLETF rats [44].
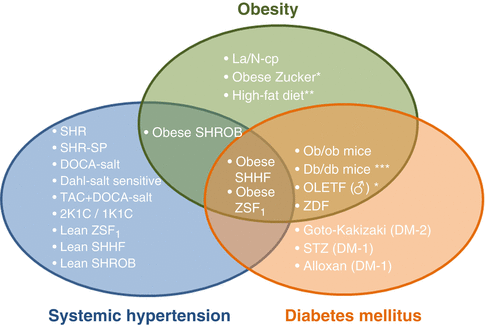
Fig. 19.2
Overview of some of the most important animal models of cardiovascular or cardiovascular-related diseases and their features. DM-1 type 1 diabetes mellitus, DM-2 type 2 diabetes mellitus
In the human setting, the development of obesity and metabolic syndrome is mostly linked with increased caloric intake and lack of physical activity, in addition to genetic predisposition. Thus it would be relevant to study the pathogenesis of CV disease resulting from abnormal food intake. Many models of obesity are induced by the consumption of modified diets, such as high-fat or high-carbohydrate diets, therefore representing potentially more relevant pathophysiological models of the human disease. Such is the case of C57BL/6 mouse fed with a high-fat diet described previously as an animal model or vascular dysfunction and atherosclerosis. Still regarding mice, ob/ob mice [45], with leptin deficiency, and db/db mice [46], presenting a mutation in leptin receptor, constitute the most used models of obesity, which also display hyperglycemia and hyperlipidemia without hypertension and thus represent a good animal model of metabolic syndrome.
Apart from these models, other genetic modifications gave rise to a whole range of transgenic including knockout and knockin mice, which are briefly described throughout Table 19.1, wherein the most usual DM animal models are also described.
19.2.3 Cardiac Hypertrophy
Cardiac hypertrophy may subside as a response of the heart to hemodynamic overload, during which terminally differentiated cardiomyocytes increase in size without undergoing cell division. This subchapter will only describe animal models of LV hypertrophy, while the subchapter regarding “pulmonary hypertension, emphysema and right ventricular failure” will address animal models of right ventricular HYP.
19.2.3.1 Pressure-Overload Induced Hypertrophy
Aortic constriction (banding) is a well-established surgical technique to induce LV chronic pressure overload and hypertrophy. Initially, aortic banding imposes almost no restriction to aortic flow but as the animal grows, the severity of the constriction gradually increases, resulting in cardiac hypertrophy. Banding in several aortic segments (ascending (AAC), transverse (TAC) or supra/infra-renal abdominal aorta) has been used to mechanically reproduce the cardiac consequences of aortic stenosis, coarctation of the aorta and systemic hypertension [47]. The timeline of the disease progression depends on the selected specie, age, gender, anatomic locations or degree of the constriction (Fig. 19.3).
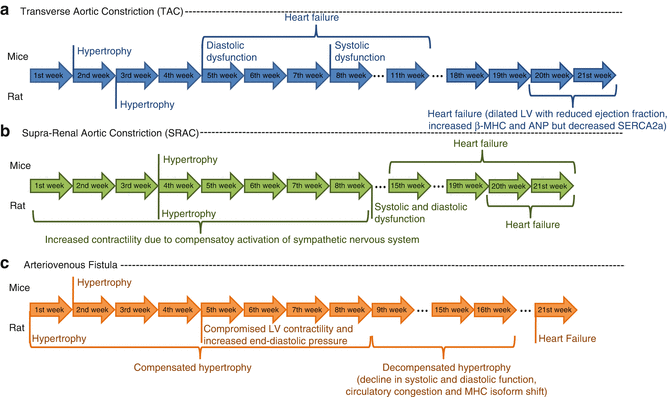
Fig. 19.3
Timeline progression of three animal models of left ventricular hypertrophy in mice and rat: Transverse aortic constriction (TAC), Supra-renal aortic constriction (SRAC) and arteriovenous fistula
Changes in nitric oxide (NO) pathway are believed to play an important role in the pressure-overloaded heart and pathological cardiac remodelling (REM). In fact, reports showed that the phosphodiesterase-5 inhibitor sildenafil reduces LV HYP and dilatation in a mouse TAC model [48]. Additionally, the exogenous administration of the NOS cofactor BH4, has been shown to reduce LV HYP, fibrosis, and cardiac dysfunction in mice with pre-established pressure-overload hypertrophy. In this setting, BH4 re-coupled endothelial NOS, with subsequent reduction of NOS-dependent oxidative stress and reversal of maladaptive remodelling [49].
Among the major advantages that these banding models share compared to other hypertensive or HF models, is the ability to manipulate the degree of pressure overload by changing the constriction severity [50]. Concerning the thoracic aorta constrictions the main advantage is the similarities to human HF progression, especially to aortic stenosis patients. Accordingly, it is characterized by initial compensatory phase, with concentric LV hypertrophy followed by an enlargement of cardiac chambers associated with a further deterioration of LV function [51]. Another advantage is the extensive information regarding TAC model: it was first described in 1994 by Rockman [52] and it has been extensively used since then, especially in mice either by traditional thoracotomy approach or by minimally invasive aortic banding through a small incision in the proximal sternum [15]. Finally, this method permits the quantification of the pressure gradient across the aortic constriction and the stratification of LV hypertrophy [13]. However, the TAC model has several drawbacks such as the prolonged duration of the protocols (Fig. 19.3), inter-individual variability in the response to pressure overload, high proportion of debanding due to internalization of the constriction knot [15]. These two last disadvantages require the use of large experimental groups and the use of accessory methods for visualization of the constriction integrity and progression of disease, such as echocardiography. Both AAC and TAC models have a common disadvantage resultant from the complex surgical method and equipment necessary for open-chest microsurgery. The lack of such an extended learning curve is the major advantage of abdominal constriction model, alongside with the lower mortality rate associated with banding (10 %). Activation of RAAS might however limit the use of abdominal aorta constriction in some studies [53]. Moreover, decrease of LV relaxation rates makes such models valuable for the evaluation of diastolic dysfunction, which is an important factor in the progression of LV failure [54]. In addition, the stimulus for HF is gradual in onset as is the progression from compensated HYP to HF in humans, thus making it clinically more relevant.
19.2.3.2 Volume-Overload Induced Hypertrophy
Arteriovenous shunts or fistulas have been used to induce volume overload, HYP and later DCM and HF in rodents. Femoral artery to femoral vein or aorta to cava fistulas lead to HF, but present a mortality rate above 25 % in all studies [55]. The more recent aortocaval shunt is a relatively simpler and a faster alternative to induce HF with good survival rates and no need to perform thoracotomy [56]. Compared to the rat, reports on the cardiac consequences of arteriovenous shunts in mice are still scarce. The progression of myocardial changes is represented in Fig. 19.3. Notably conflicting observations have been reported probably due to differences in the techniques utilized to induce volume overload, duration and size of the shunt or even the stage of cardiac hypertrophy. This procedure has the advantage of being fast and usually well tolerated, despite the limitation of requiring a laparotomy. Nonetheless, shunt closure has been reported in 7 % of the cases, which means that it is necessary to confirm the patency of the shunt at the end of investigation.
Another procedure used to induce volume overload in rats is aortic valve regurgitation, in which an aortic valve cusp is punctured [57].
19.2.4 Dilated Cardiomyopathy
Engineered mice with mutations or lacking certain cytoskeleton genes are known to cause dilated cardiomyopathy (DCM). Such is the case of dystrophin and utrophin knockout mice that develop severe muscular dystrophies and cardiomyopathy similar to humans. Also knockout mice for muscle LIM protein, melusin, tumor necrosis factor-α (TNF-α), protein kinase C-β2 (PKC-β2) and calmodulin kinase II display phenotypic features of the human DCM (Table 19.2) [58]. Besides genetically modified mice only a few animal models are able to satisfactorily mimic DCM, such as SHHF/Mcp-fa cp which, regardless of the gender or genotype, eventually develop spontaneous DCM accompanied by subcutaneous edema, dyspnea, cyanosis, lethargy, enlarged hearts, thickened ventricles, dilated heart chamber, ascites, pulmonary edema and pleural effusion. Moreover, several animal models used for other CV diseases developed DCM, that is, myocarditis or myocardial ischemia. Apart from rodents, DCM can be induced by chronic rapid pacing at a frequency three- to fourfold higher than the spontaneous heart rate and is mostly applied to dogs, but also to pigs, sheep and monkeys [58].
19.2.4.1 Doxorubicin
Doxorubicin (DOX) is an anthracycline widely used in cytostatic treatments. One of the major long-term consequences of DOX therapy is the development of dose-dependent cardiomyopathy and ultimately congestive HF either in humans as in various animal species. Therefore, understanding the pathogenesis of cardiotoxic cardiomyopathy is essential to the development of new measures to prevent cardiotoxicity associated with antineoplastic therapies [15].
DOX administration once a week for 6 weeks or on alternate days for 2 weeks has been shown to induce cardiomyopathy and HF in rodent, while in rabbits the first abnormalities are observed after 3 months’ injections twice weekly. Interestingly, a single intravenous administration of DOX has been shown to induce significant LV dysfunction in mice after 5 days, while its intracoronary injection allows a smaller dose of DOX to induce HF without systemic toxicity [15].
DOX-induced cardiomyopathy is characterized by ventricular wall thinning and dilatation (reduced mass-to-volume ratio), and depressed systolic and diastolic function accompanied by fluid retention and by neurohumoral activation. At the cardiac muscle level, DOX promotes intrinsic contractile dysfunction and reduced contractile reserve. Furthermore, DOX impairs vascular, as well as cardiac endothelial function and induces inflammatory reactions in the heart, leading to thrombosis in the atria and myocarditis [15].
Multiple pathways of antracycline-induced cardiac cellular injury have been proposed, such as the release of cardiotoxic substances which subsequently accumulate in cardiomyocytes, the generation of free radical, lipid peroxidation and suppression of DNA, RNA and protein synthesis. Other studies suggest that cardiotoxicity pathways include abnormalities in Ca2+ handling; induction of mitochondrial DNA lesions; degradation of myofilamental and cytoskeletal proteins, including titin and dystrophin; interference with various pro-survival kinases [59] and changes in adrenergic and adenylate cyclase function. All these mechanisms may contribute to cardiac cell damage, ultimately leading to myocyte death, either by necrosis or by apoptosis [15].
DOX model has a short time course of induction of HF and also the advantages of being technically simple, reproducible, non-invasive, and economical. Additionally, it can be used in several animal species to promote either chronic or acute HF [60]. The main limitations of this model are related to the variable degree of ventricular dysfunction and the high incidence of arrhythmias that contributes to the high mortality rate. Importantly, it is known that doxorubicin cardiotoxicity is dose-dependent and may appear either during and within 2–3 days of its administration or later within 30 days of administration of its last dose. In humans, it may occur even after 6–10 years after its administration [61]. Anthracycline administration also has undesirable bone marrow, gastrointestinal and renal toxicity that can however be avoided by the intracoronary injection of the drug [60
Table 19.2
Brief description of the most important genetically engineered mouse models used for heart failure research
Gene | Mechanism | Structural changes | Functional changes | Comments | ||
|---|---|---|---|---|---|---|
Cytoskeletal and sarcomeric proteins | Melusin [62] | Knockout | Cardiac chamber dilation and relative wall thinning | Depressed systolic function and myocardial contractility, pulmonary congestion and impaired adrenal responsiveness | Melusin is a muscle specific integrin β1-interacting protein. Probably required for LVH induced by biochemical stress but not by humoral stimulation | |
No effect on cardiac structure or basal function in physiologic conditions or in response to ATII or phenylephrine | ||||||
Leads to LVH and favours the transition toward dilated cardiomyopathy and contractile dysfunction in response to chronic pressure overload | ||||||
Muscle LIM protein (MLP) [63] | Knockout | Myofiber disarray, severe LV hypertrophy and dilation and fibrosis | Systolic dysfunction and CHF | Suitable model for long-term observation of drug effects on cardiac function and remodelling (near normal life span). Mutations in MLP were identified in patients with DCM or hypertrophic cardiomyopathy [64] | ||
Blunted response to β-agonist stimulation | ||||||
Proteins linked to FHC (β-MHC, troponin T, MyBP-C) [65] | Knockout | These models are important to study the mechanisms linking the primary mutation and the secondary development of CHF. Moreover, environmental or genetic factors play a role in the development of each phenotype | ||||
Proteins linked to heritable dilated cardiomyopathy (actin, β-MHC, troponin T) [66] | Desmin | Knockout | Severe disorganization of muscle architecture associated with degeneration [67] | Mutation in β-cristallin (a molecular chaperone heat shock protein essential for cytoskeletal integrity) causes a desmin-related myopathy [68] | ||
Cardiac actin | Reduced number of actin filaments and myofiber disarray | Early postnatal death [69] | ||||
Dystrophin | DCM only in the presence of concomitant urotrophin deficiency [70] | |||||
Neurohumoral receptors | Sympathetic nervous system | Mutation | Ventricular hypertrophy increased ANF expression (sufficient for hypertrophy development) | Clinical studies with α1-AR selective blockers or activators of central α2-ARs have so far provided disappoint results [73] | ||
The activation of subtype-specific α2-AR at peripheral nerves has not been tried yet [74] | ||||||
α2A or α2C-AR [75] | Knockout | Faster progression to CHF and death when challenged with pressure overload (TAC) than wild-type or α2B knockout mice | Feedback pre-synaptic inhibition seems to be important to attenuate the detrimental effect of catecholamines | |||
β1-AR | Significant hypertrophy of the myocytes, altered nuclear morphology and concomitant fibrosis, with increased expression of apoptotic proteins in fibrosis areas | Elevated basal heart frequency and increased contractility in young animals; at 35 weeks of age the contractility decreases and many animals present clinical signs of HF and die before the age of 14 months. Contractile dysfunction is preceded by impaired calcium handing [78] | Chronic activation clearly implicated in CHF development in both animal and clinical studies | |||
In a model with concomitant deletion of PLB gene, mice did not die prematurely anymore, the cardiac hypertrophy and fibrosis were inhibited, LV function was preserved and also the disturbed Ca2+ handling was restored. These findings show that the β1-adrenoceptor–PLB-signalling cascade is a crucial regulator of cardiac contractility [79, 80] | ||||||
β2-AR | Overexpression [81] | In CHF there is selective down-regulation of β1-AR and desensitization of both β1-AR and β2-AR (mediated by β-ARK1) | ||||
Mice subjected to pressure overload through aortic constriction showed higher mortality and incidence of heart failure | ||||||
β-ARK1 [93] | Knockout | Mice do not survive | ||||
β-ARK peptide inhibitor [93] | Overexpression | Increased cardiac contractility | Cross-breeding with cardiomyopathic MLP-knockout mice restores the responsiveness to β-agonist stimulation and enhances cardiac contractility [94] | |||
Cross-breeding with mice overexpressing calsequestrin enhances cardiac function and reduces mortality. Combination with the β-blocker metoprolol has an additive effect [95]. β-ARK1 seems to be a putative therapeutic target | ||||||
Neurohumoral receptors | Renin-angiotensin system | AT-1 [96] | Overexpression | Significant cardiac hypertrophy and remodelling with increased expression of ANF and interstitial collagen deposition | CHF in the absence of blood pressure changes | This model attests the direct role of AII in cardiac hypertrophy |
Cytokines | Overexpression | Biventricular dilation, fibrosis, interstitial infiltrates and hypertrophy | Marked decline in cardiac function and blunted responsiveness to β-agonist stimulation | |||
Overexpression | Ventricular hypertrophy | No progression to CHF | Gp130 signalling cascade could be a potential therapeutic target because of its cardioprotective effect | |||
Disruption | Myocardial apoptosis | Accelerated transition to CHF when subjected to pressure overload | ||||
Cellular signalling proteins | Heterotrimeric G proteins | Gαq | Overexpression [102] | Cardiac hypertrophy with increased expression of ANF and β-MHC | Blunted response to β-agonist stimulation | Gαq signalling is necessary and sufficient to the development of cardiac hypertrophy |
Modest | Mice with attenuated hypertrophic response to pressure overload (expression of Gαq peptide inhibitor or lack of norepinephrine) have increased wall stress but no decline in cardiac function [103] | |||||
High | Overt CHF | |||||
Transient expression of constitutively active Gαq [104] | Cardiac hypertrophy | Dilated cardiomyopathy | Gq signalling pathway could be an interesting therapeutic target to prevent cardiac hypertrophy and CHF | |||
Resistance to hypertrophy induced by pressure overload | ||||||
Gαs | Modest overexpression [107] | Cardiomyopathy similar to that induced by chronic catecholamine administration in elderly mice (cellular degeneration, necrosis, replacement fibrosis) | ||||
Gi | Conditional expression of Gi-coupled receptor (R01) [108] | High-level of expression causes hyperactive Gi signalling with systolic dysfunction. Suppression of R01 expression after 8 weeks prevents further myocardial degeneration and partially improves systolic function and CHF | It is still matter of controversy if up-regulation of Gi pathway is a compensatory response to hyperactive Gs signalling | |||
Cellular signalling proteins | Small GTPases | Ras [109] | Ventricular expression of a constitutively active form | Hypertrophy | Diastolic dysfunction | |
RhoA [110] | Overexpression | Sinus and atrioventricular nodal dysfunction and contractile failure without ventricular hypertrophy | ||||
Rac1 [111] | Myocardial expression of constitutively active form | Transient hypertrophy resolving with age | Lethal dilated cardiomyopathy | |||
Protein kinases | PKC-β2 [112] | Overexpression | Cardiac dilation and fibrosis | Reduced cardiac function | ||
PKC-α [113] | Knockout | Less systolic dysfunction and LV dilation in response to LAD | Cardiomyocyte-specific expression of a PKC-α inhibitor leads to the same results of knockout [114] | |||
PKC-ε [115] | Overexpression | Mild concentric hypertrophy without fibrosis | Normal cardiac function | |||
PKA [116] | Expression of catalytic subunit in heart | Dilated cardiomyopathy with arrhythmias and reduced cardiac function | Enhanced Ca release from SR in response to β-AR signalling | |||
Calmodulin kinase II [117] | Overexpression | Progressive LV hypertrophy and dilation | Overt CHF | CaMKII contributes to heart failure progression. | ||
Expression of a cardiomyocyte-specific CaMKII inhibitory peptide reduces LV hypertrophy and dilation and systolic dysfunction. [118] A pharmacological inhibitor of CaMKII (KN93) attenuated LV dilation and improved systolic function post-MI in wild-type rats suggesting that this kinase could be a potential interesting therapeutic target [119] | ||||||
Calmodulin [120] | Overexpression | Myocardial hypertrophy | No CHF | Other downstream pathways of calmodulin signalling could prevent the progression to CHF | ||
Calcineurin [113] | Overexpression | Myocardial hypertrophy | CHF development within 2 months | |||
Calcium-regulating proteins | L-type Ca channels [121] | Overexpression | Ventricular hypertrophy, apoptosis and fibrosis | Overt CHF within 8 months and death of CHF within 12 months in the majority of mice < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||