Fig. 1.1
Distinct pharmacological approach to bock the renin-angiotensin system (RAS)
AT1 receptors belong to the superfamily of G-protein–coupled receptors that contain seven trans-membrane regions and are localized in the kidney, heart, vascular smooth muscle cells, brain, adrenal gland, platelets, adipocytes, and placenta. The AT1 receptor mediates most of the detrimental effects of Ang II on cardiovascular system. In particular, their stimulation induces vasoconstriction, increases Na+ retention, and endothelin secretion, enhances vasopressin release, activates sympathetic nervous activity, promotes cardiomyocytes hypertrophy, stimulates vascular and cardiac fibrosis, increases myocardial contractility, induces arrhythmias, stimulates plasminogen activator inhibitor 1, and stimulates superanoxide formation. ARBs act by blocking the AT1 receptors and thus, prevent the pathophysiological effects mediated by the binding of Ang II to the AT1 receptor. Moreover, as a consequence of AT1 blockade, ARBs increase Ang II levels above baseline. Increased plasma levels of Ang II result in unopposed stimulation of the AT2 receptors. It has been proposed that stimulation of AT2 receptors exerts an important role in counterbalancing some of the detrimental effects of Ang II mediated by AT1 receptors, such as inhibition of cell growth, promotion of cell differentiation, and synthesis of NO. Finally, for some ARBs has been documented an agonist action on PPAR-γ receptors. These biological effects are translated in different pharmacological actions such as prevention of hypertension-induced TOD, of hypertension-associated diseases such as diabetes, atherosclerosis, and renal disease; interestingly, these effects appear to be potentially independent from ARBs-induced BP reduction.
RAS Inhibition and Blood Pressure Lowering
The relation between BP and CV events is a continuous phenomenon without threshold [52] The RAS plays a pivotal role in the regulation of BP; therefore, drugs affecting RAS like ACE and ARBs are largely used for BP control and for management of CV risk. The CAPPP study was the first prospective, randomised intervention trial aimed to evaluate the effects of ACE inhibition and conventional therapy on cardiovascular morbidity and mortality in patients with essential hypertension. This study enrolled 10,985 patients: 5,492 patients assigned captopril and 5,493 assigned conventional therapy; the two treatment regimens had the same effect on blood pressure, and incidence of major cardiovascular events [30]. The interpretation of the results of this study was that an antihypertensive regimen based on ACE inhibitors was as effective as conventional treatment with diuretics, β-blockers, or both in prevention of cardiovascular morbidity and mortality. Similarly the STOP-2 study [29] showed in elderly patients that the incidence of cardiovascular events was similar in elderly hypertensives randomized to a calcium antagonist, an ACE inhibitor, or conventional treatment with a diuretic or a β-blocker, and decrease in blood pressure was of major importance for the prevention of cardiovascular events. ARBs represent a class of effective and well tolerated orally active antihypertensive drugs. The antihypertensive effects of the ARBs have been demonstrated by many interventional studies. In a meta-analysis Conlin et al. analyzed 43 randomized, controlled clinical trials comparing the antihypertensive effects of ARBs (losartan, valsartan, irbesartan, and candesartan) with placebo, other antihypertensive classes, and each other. The analysis included 11,281 patients treated with ARBs showed that the weighted average of diastolic and systolic blood pressure (BP) reductions and responder rates among agents were comparable, irrespective of starting doses, monotherapy dose titration, and combination therapy with hydrochlorothiazide (HCTZ) [15]. Available data indicate that both ACE-inhibitors and ARBs are comparable to other antihypertensive agents in lowering blood pressure: However, these two classes of drugs are more effective, compared to others antihypertensive drugs, in the prevention of metabolic abnormalities and sub-clinical organ damage.
RAS Inhibition and Hypertension-Related Metabolic Abnormalities
Essential hypertension often is associated with metabolic abnormalities which can exert a detrimental effects on the prognosis. Therefore, the prevention of diabetes, metabolic syndrome and dyslipidemias represents a key point of antihypertensive treatment. Inhibition of RAS has been demonstrate to have a favorable effect in the prevention of such abnormalities.
Dyslipidemias
Clinical evidence: Interventional studies have demonstrated that pharmacological interference of RAS, slightly improves the lipid profile in hypertensive patients. This beneficial action has been demonstrated for the different ARBs. In particular, Kyvelou et al. demonstrated, in a cohort of 2,438 hypertensive patients, followed for 6 months, that treatment with ARBs-based monotherapy induce a significant reduction of total and LDL-cholesterol, in addition increase HDL-cholesterol [50]. Furthermore, a sub-study of LIFE showed in hypertensive patients that, in comparison with atenolol-based, losartan-based regimen, induces a less decrease in HDL-C; and this pharmacological effect is associated with a better prognosis [66]. The authors speculated that less decrease in HDL-C may explain around one-third of the beneficial effect of losartan-based compared with atenolol-based antihypertensive treatment on composite end-point found in the LIFE study. The favorable effects on lipid profile have been documented for ARBs also when these are combined with antihypertensive drugs that aggravated metabolic profile. In particular, the Alpine Study showed that treatment with diuretics, if needed, in combination with a β-blocker was associated with a worsening of metabolic profile; this effect was not detected for patients treated with an ARB [55]. Some ARBs such as telmisartan and eprosartan, have been reported to stimulate peroxisome proliferator-activated receptor-γ (PPARγ) [82] and could thus improve insulin sensitivity [67]. This, indirectly could influence systemic lipid concentrations, so it is unclear whether the differences in results from clinical trials arise from these ancillary properties of RAS blockade. However, two studies failed to demonstrate the beneficial effects of ARBs on lipid profile in hypertensive patients. In particular Grassi et al. in the CROSS study evaluated in obese hypertensive individuals the antihypertensive, neuroadrenergic, and metabolic effects of an ARB in comparison with a diuretic. The results of this study showed that after 3 months of treatment, despite Candesartan improved insulin sensitivity had no effect on plasma levels of triglycerides, of HDL-cholesterol and of LDL-cholesterol [27]. More recently, Nishida et al. confirmed the observation of Grassi, demonstrating that in patients with mild to moderate hypertension Candesartan has no effect on plasma levels of triglycerides, of total and of LDL-cholesterol [65].
Experimental evidence: ANG II exerts several effects that influence atherogenic properties of cholesterol. In particular, it has been demonstrated that AT1A receptor deficiency had a striking effect in reducing hypercholesterolemia-induced atherosclerosis in LDL receptor-negative mice [19]. Moreover, in this model hypercholesterolemia was associated with increased systemic angiotensinogen and angiotensin peptides, which were reduced in AT1A receptor-deficient mice, suggesting that LDL cholesterol contributes to development of atherosclerosis through a RAS-dependent mechanism. Moreover, it has been reported in primary cultures of human monocyte-macrophages, that the pro-atherogenic effects of ANG II are related to the property of ANG II to upregulate the expression of Acyl-CoA: cholesterol acyltransferase-1 (ACAT1) [41]. This enzyme converts free cholesterol into esters for storage in lipid droplets. This process could promote foam cell formation, and increase cholesterol content of atherosclerotic lesions. Finally, Several studies have reported that ANG II increased the oxidation of LDL in macrophage cell lines as well as mouse peritoneal macrophages, possibly through activation of NADPH oxidase [42]. Altogether these observations are consistent with the notion that ANG II may influence the atherogenic properties of cholesterol without necessarily changing the blood concentrations [69]. Interestingly, cholesterol is capable to regulate RAS. In particular, it has been demonstrated the capability of LDL cholesterol to increase AT1 receptor gene expression on vascular smooth muscle cells [64] as well as, oxidized LDL can also increase AT1 receptor gene expression in human coronary artery endothelial cells [54]. Together, these results clearly demonstrate a cross-talk between hypercholesterolemia and RAS in the development of atherosclerosis.
Metabolic Syndrome and Obesity
Clinical evidence: Metabolic syndrome (MetS) has been defined in different ways, and it is a risk factor for development of atherosclerosis and occurrence of CV events. A feature of MetS is the constellation of risk factors including abdominal adiposity, impaired fasting glucose, hypertension, and dyslipidemia. Moreover, obesity also predisposes to CV disease and often is associated with other abnormalities of the MetS. In particular, adipose tissue acts as an endocrine organ, secreting hormones and other substances that create a proinflammatory state and promote formation of atherosclerotic plaques [51]. In the last years many interventional studies specifically addressed the effects of RAS inhibition/antagonism in metabolic syndrome. In particular the hemodynamic and metabolic effects of two ARBs were particularly evaluated: telmisartan and irbesartan. These molecules activates effect on the activity of peroxisome proliferator-activated receptor gamma, a well-known target for insulin-sensitizing antidiabetic drugs. In particular, the ISLAND [87] study demonstrated that Administration of irbesartan and/or lipoic acid to patients with the metabolic syndrome improves endothelial function and reduces proinflammatory markers, factors that are implicated in the pathogenesis of atherosclerosis The OLAS study evaluated the effects of different treatments with olmesartan/amlodipine and olmesartan/hydrochlorothiazide on inflammatory and metabolic parameters in non-diabetic hypertensive patients with MetS [60]. This study showed that olmesartan-based combinations were effective, but the amlodipine combination resulted in metabolic and anti-inflammatory effects that may have advantages over the hydrochlorothiazide combination. More recently, it was demonstrated the capability of telmisartan to activates PPARγ in circulating monocytes of patients with the metabolic syndrome [6]. Many intervention trials have not been designed specifically for obese hypertensive patients, and only few studies have specifically addressed the use of ACE-inhibitors or ARBs in these patients. For instance, the TROPHY study, was a multicenter, double-blind trial that evaluated the efficacy and safety of the lisinopril, against the hydrochlorothiazide, in obese, hypertensive patients [71]. The results of this study showed that, despite similar reductions in office systolic BP and diastolic BP with lisinopril or hydrochlorothiazide, treatment with angiotensin-converting enzyme inhibitors has a greater efficacy as monotherapy at lower doses compared with thiazide diuretics.
Experimental evidence: Several mechanisms account for the association between MetS and increased risk of atherosclerotic CV events. For instance, there is growing evidence that RAS, through Ang II, is involved not only in the pathogenesis of hypertension and atherosclerosis, but also plays a role in the development of insulin resistance. Moreover, it has also demonstrated that activation of the RAS in adipose tissue represents an important mechanism that account for the link between obesity and hypertension [20]. Adipose tissue is an important production site of angiotensinogen, and it has been reported a correlation between plasma level of angiotensinogen, blood pressure, and body mass index [74]. Moreover, in obese Zucker rats it has been documented an increase higher than 50 % of gene expression of angiotensinogen, in adipose tissue compared with lean rats [39]. Interestingly, it has been also demonstrated that Ang II is implicated in the regulation of lipid synthesis and storage in the adipocytes [20], as well as, in adipocyte growth and differentiation [2]. In addition, it has been documented that the AT1 receptor, and ACE genes were found to be upregulated in the adipose tissue of hypertensive patients with obesity [25]. Altogether these experimental evidence suggest a strong relationship between RAS and regulation of functional activity of adipose tissue, this phenomenon could be involved in the increase of both BP and CV risk.
Insulin Resistance and Diabetes
Clinical evidence: The beneficial effects of ACE inhibitors to improve insulin resistance are also evident from several observational and interventional studies in human subjects with hypertension and type 2 diabetes. For instance, in the Captopril Prevention Project (CAPPP) [30] and the Heart Outcomes Prevention Evaluation (HOPE) study [105], two large prospective studies involving hypertensive subjects at risk for developing type 2 diabetes, there was a lesser incidence of newly-diagnosed type 2 diabetes in those subjects who received an ACE inhibitor (either captopril or ramipril) compared to the respective placebo control groups. More consistently, in 2005, it has been published a meta-analysis of 12 randomized controlled clinical trials of ACE inhibitors or ARBs to evaluate the efficacy of these medications in diabetes prevention. This meta-analysis, involving 72,333 non-diabetic patients (approximately 338,000 patient-years of follow-up), with mean duration of follow-up ranged from 1 to 6.1 years, showed that ACE inhibitors or ARBs produced a highly significant 25 % reduction (or a decrease from 17.4 to 14.3 cases per 1,000 patient-years) in the incidence of new-onset diabetes [1]. The clinical implications of this meta-analysis are important because the development of diabetes is associated with insulin resistance. Therefore, it is possible to speculate that the inhibition of RAS by both ACE inhibitors or by AT 1 receptor antagonists ameliorates insulin sensitivity (Fig. 1.2). The mechanisms of action whereby these classes of drugs improve insulin sensitivity are complex and multifactorial. In particular, ACE inhibitors not only block the conversion of angiotensin I to angiotensin II, but also increase bradykinin levels through inhibition of kininase II-mediated degradation. The higher kinin levels lead to an increased production of prostaglandins (prostaglandin E1 and prostaglandin E2) and nitric oxide, which improve exercise-induced glucose metabolism and muscle sensitivity to insulin [24], resulting in enhanced insulin-mediated glucose uptake. Furthermore, the peripheral vasodilatory actions of ACE inhibitors and ARBs lead to an improvement in skeletal muscle blood flow, the primary target for insulin action and an important determinant of glucose uptake. This effectively increases the surface area for glucose exchange between the vascular bed and skeletal muscles. Clinical evidence supporting this effect has been provided by Morel et al. [61], who have demonstrated improved insulin sensitivity when enalapril was given for 12 weeks to 14 obese, hypertensive, and dyslipidemic patients. A similar effect has also been reported with captopril [68]. However, it should be underlined that this action can not be considered the main mechanism that account for the increase of insulin sensitivity because this effect is not shared by drugs that acts as vasodilators like hydralazine. Moreover, the protection against the development of insulin resistance may be partially due to a regulation of adipocyte function. It has been demonstrated that increased levels of Ang II inhibit pre-adipocyte differentiation into mature adipocytes, and this impairs the fat cells’ ability to store fat. This, in turn results in shunting of fats to the liver, skeletal muscle, and pancreas, which worsens insulin resistance. Reducing Ang II levels with an ACE inhibitor or blocking the angiotensin II receptor type 1 with an ARB may promote differentiation of pre-adipocytes to mature adipocytes, which serve as a sump for fat. In addition, redistribution of the lipids from the peripheral tissues would improve insulin sensitivity [84]. Another mechanism that accounts for the favorable effect of ARBs and ACE inhibitors on insulin sensitivity relates to a possible protective effect on the pancreatic beta cell, through inhibiting the vasoconstrictive effect of angiotensin II in the pancreas and increasing islet blood flow [10], which could improve insulin release by beta cells. Telmisartan, an ARB, has been shown to act as a peroxisome proliferator-activated receptor (PPAR)-gamma agonist, similar to the thiazolidinediones rosiglitazone and pioglitazone, which preserve pancreatic beta-cell function [82]. Peroxisome proliferator–activated receptor γ is a transcription factor that controls the gene expression of several key enzymes of glucose metabolism and thereby increases insulin sensitivity.
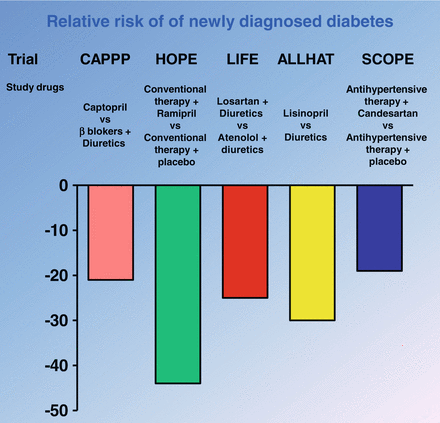
Fig. 1.2
Results of principal intervention trial with renin-angiotensin system (RAS) inhibitors on new diagnosis of diabetes
Experimental and clinical studies suggest that blocking the effects of Ang II (through ACE inhibition or ARBs) increases insulin sensitivity, skeletal muscle glucose transport, and pancreatic blood flow, which may contribute to the prevention of diabetes mellitus. Therefore, ACE inhibitors or ARBs represent the logical first-line anti-hypertensive agent in patients with impaired fasting glucose or metabolic syndrome for multiple reasons, including the reduction in risk of progression to overt type 2 diabetes. Even in patients without diabetes or metabolic syndrome, what was previously thought to be a “high-normal” blood pressure (≥130/80 to 139/89 mmHg) is associated with an increased risk of adverse cardiovascular events [100].
Experimental evidence: In vivo and in vitro studies have shown that Ang II stimulation also induces insulin resistance. Overactivity of the RAS observed in cardiovascular diseases is likely to impair insulin signaling and contribute to insulin resistance. Actually, Ang II acting through the AT1 receptor inhibits the actions of insulin in vascular tissue, in part, by interfering with insulin signaling through PI3K and downstream Akt signaling pathways via generation of reactive oxygen species (ROS) by NADPH oxidase. ROS are important intracellular second messengers that activate many downstream signaling molecules, such as phosphotyrosine phosphatases (PTPase) and protein tyrosine kinases. PTPases are critical regulators of tyrosine phosphorylation-dependent signaling, and tyrosine dephosphorylation by PTPases may play a crucial role in Ang II-induced insulin resistance. Several PTPs have been implicated in the Ang II-induced dephosphorylation of insulin receptor. However, the most convincing data, support a critical role for PTP-1B in insulin action. Actually, PTP-1B knockout mice display increased insulin sensitivity and maintain euglycemia (in the fed state) with one-half the level of insulin observed in wild-type littermates. Interestingly, these mice are also resistant to diet-induced obesity when fed a high-fat diet.
Recent studies have demonstrated that the generation of ROS is implicated in the Ang II-induced insulin resistance. In this regard it has been demonstrated that in vascular smooth muscle cells isolated from rat thoracic aorta Ang II profoundly decreases IRS-1 protein levels via ROS-mediated phosphorylation of IRS-1 on Ser307 and subsequent proteasome-dependent degradation. The key role of ROS in the pathogenesis of Ang II-induced insulin resistance has been also confirmed by in vivo studies. In particular, in rats chronic infusion of Ang II reduced insulin-induced glucose uptake during hyperinsulinemic-euglycemic clamp, and increased plasma cholesterylester hydroperoxide levels, indicating an increased oxidative stress. Treatment with tempol, a superoxide dismutase mimetic, normalized plasma cholesterylester hydroperoxide levels in AII-infused rats. In addition, tempol normalized insulin resistance in AII-infused rats, as well as enhanced insulin-induced PI 3-kinase activation, suggesting that Ang II-induced insulin resistance can be restored by removing the oxidative stress. On the other hand, in the endothelial cells, Ang II induces insulin resistance through the phosphorylation of IRS-1 at Ser312 and Ser616 via JNK- and ERK1/2-dependent mechanisms, respectively. This impairs the interaction of IRS-1 with the p85 regulatory subunit of PI 3-kinase and compromises the insulin vasodilatory signaling pathway involving PI 3-kinase/Akt/eNOS. Altogether, these observations provide clear insight into the mechanisms of Ang II–induced insulin resistance. ACE inhibitors decrease the conversion of angiotensin I to Ang II, in addition, via the inhibition of the kininase II breakdown, enhance the circulating level of the bradykinin (Fig. 1.3). There are several experimental data indicating that these changes in Ang II and bradykinin improve skeletal muscle glucose metabolism. In particular, there are experimental studies performed in animal models of hypertension and insulin resistance which have demonstrated that acute and chronic administration of ACE inhibitors can improve insulin action on whole-body and skeletal muscle glucose disposal. In particular, it has been demonstrated in 20-month-old rats that the acute oral administration of the ACE inhibitor captopril enhances whole-body insulin action on glucose disappearance during an intravenous insulin tolerance test by modulating the early steps of insulin signaling, and that this effect may be simulated by the administration of bradykinin [11]. Moreover, the acute infusion of captopril to obese Zucker rats, a rodent model of insulin resistance, glucose intolerance, and dyslipidemia, enhances insulin sensitivity during a euglycemic, hyperinsulinemic clamp [4]. A similar response to an acute captopril infusion has been observed in an insulin resistant diabetic dog model using this euglycemic, hyperinsulinemic clamp technique [98]. In addition to acute administration, also chronic treatment with ACE inhibitors has been demonstrated to enhance glucose tolerance in animal models of insulin resistance. In fact, it has been shown that chronic administration of ACE inhibitors to obese Zucker rats elicits an increase in whole-body insulin action. Chronic oral treatment of obese Zucker rats with the ACE inhibitors captopril [18], or trandolapril [89] causes substantial improvements in whole-body insulin sensitivity, assessed during an oral glucose tolerance test, and these ACE inhibitor-mediated improvements in whole-body insulin sensitivity are associated with decreases in plasma insulin and amelioration of dyslipidemia. Chronic administration of an ACE inhibitor to a mouse model of type 2 diabetes (KK-Ay) also significantly improves whole-body glucose tolerance and insulin sensitivity [85].
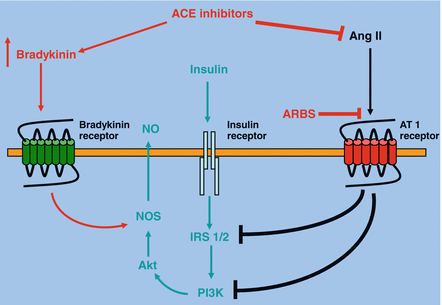
Fig. 1.3
Molecular mechanisms that account for improvement of insulin sensitivity following renin-angiotensin system inhibition
Skeletal muscle is an important locus of ACE inhibitor action in rodent models of insulin resistance and hypertension. Acute in vivo administration of the ACE inhibitors captopril [31] or trandolapril [36] significantly enhances insulin-mediated glucose transport activity in skeletal muscle in the obese Zucker rat. Skeletal muscle is not the only muscle tissue in which ACE inhibitors can beneficially modulate glucose metabolism. Rett et al. [72] have demonstrated using the perfused Langendorff preparation that the acute administration of active ACE inhibitor metabolite phosphorylate can significantly increase insulin-stimulated glucose transport activity in cardiac muscle of the obese Zucker rat, and that this effect can be mimicked by administration of bradykinin.
RAS Inhibition and Hypertension-Related TOD
Subclinical organ damage is an important determinant of CV risk in essential hypertension, and represents a key target of antihypertensive therapy. Interestingly, Ang II plays a critical role in the pathogenesis of TOD (Fig. 1.4). Thus, RAS blockade should be considered as first choice therapy of hypertensive patients with evidence of TOD.
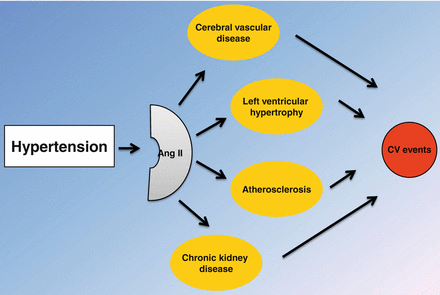
Fig. 1.4
Central role of angiotensin II (Ang II) in the pathogenesis of target organ damage and cardiovascular (CV) events in essential hypertension
Left Ventricular Hypertrophy
Clinical evidence: Left ventricular hypertrophy (LVH) is an independent risk factor for morbidity and mortality for CV diseases. BP is an important determinant of LVH, and a substantial percentage of patients with hypertension develop this complication. Several studies have analyzed the effects of different classes of antihypertensive drugs on LVH. The first meta-analysis aimed to assess the ability of various antihypertensive agents to reduce left ventricular hypertrophy was published in 1996 by Schmieder et al. [81]. This analysis considered 39 double-blind, randomized, controlled clinical studies with parallel-group design. After adjustment for different durations of treatment, left ventricular mass decreased by 13 % with ACE inhibitors, 9 % with calcium channel blockers, 6 % with beta-blockers, and 7 % with diuretics. There was a significant difference between drug classes (P < .01): ACE inhibitors reduced left ventricular mass more than beta-blockers (significant, P < .05) and diuretics (tendency, P = .08), suggesting the ACE inhibitors as first-line drugs to reduce LVH. In 2003 was published a further meta-analysis that added also ARBs-based clinical trials. This analysis considered a total 80 studies with more than 4,000 patients [44]. The principal finding of this analysis was that ARBs, calcium antagonists, and ACE-inhibitors were the most effective drug classes for reducing left ventricular mass in patients with essential hypertension (Fig. 1.5). In 2009 a metaregression-analysis assessed, in patients with essential hypertension, the predictor of the regression of LVH among the principal classes of antihypertensive drugs [21]. This analysis considered of 75 prospective, randomized comparative trials, including 6,001 patients, with a median study duration of 6 months. The main result of this analysis showed that ARBs induce larger regression of LVH. These studies indicate that RAS inhibition with both ACE-inhibitors or ARBs represents a valid pharmacologic strategy to prevent or reduce LVH.
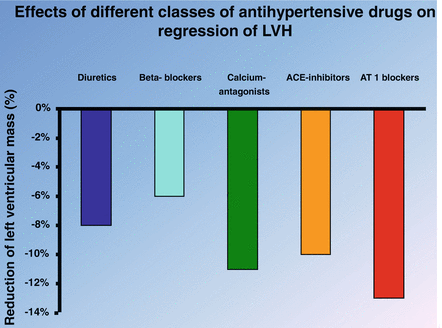
Fig. 1.5
Effects of different classes of antihypertensive drugs on regression of left ventricular hypertrophy (LVH)
Experimental evidence: Several experimental evidence suggests that Ang II has a key role in the pathogenesis of load-induced LVH. For instance, treatment of rats having aortic coarctation with an ACE inhibitor [9] or ARBs [46] prevents or causes regression of LVH. Moreover, ACE inhibitor administration also prolongs survival of rats with pressure overload [103]. These results are consistent with the involvement of the RAS in genesis of LVH and its activation by hemodynamic loading in vivo. The role of Ang II as a critical mediator of stretch-induced hypertrophy has been shown in the neonatal rat cardiac myocyte system in vitro [77]. Ang II receptor antagonists such as [Sar1 Ile8]Ang II (antagonist for the Ang II type I and II receptors) and losartan and TCV11974 (antagonists for the Ang II type I receptor) inhibit major markers of stretch-induced hypertrophy [104], which suggests that Ang II plays a critical role in stretch-induced hypertrophy in the neonatal rat myocyte culture system [79]. Several lines of in vivo evidence suggest that the cardiac RAS is upregulated chronically in load-induced hypertrophy. mRNA expression of angiotensinogen, renin, ACE, and Ang II receptor are all upregulated in cardiac hypertrophy caused by pressure overload and ischemia [93]. The upregulation of the cardiac RAS was also observed in mechanical stretch of neonatal rat cardiac myocytes in vitro [79]. Treatment of cultured cardiac myocytes with exogenous Ang II also upregulates mRNA expression of angiotensinogen, renin, and ACE, but not Ang II receptor [86]. This suggests that mechanical stretch initially causes acute secretion of preformed Ang II and that secreted Ang II may initiate a positive feedback mechanism, thereby upregulating the local renin-angiotensin system over time. It is likely, however, that upregulation of the Ang II receptor by mechanical stretch is mediated by an Ang II-independent mechanism [78]. Further studies have demonstrated the molecular mechanism that account for Ang II-mediated development of LVH. In particular, Bendall et al. demonstrated, in transgenic mice lacking the gp91phox subunit of NADPH oxidase, that 2 week-stimulation of subpressor doses of Ang II stimulation failed to induce LVH, this was associated with inhibition to superoxide production [7]. The result of this study indicated that oxidative stress is centrally involved in the direct cardiac hypertrophic response to Ang II.
Chronic Kidney Disease
Clinical evidence: Development of chronic kidney disease (CKD) a common feature of essential hypertension. The renal damage, is characterized by a progressive loss of renal function, and, at the same time, increases cardiovascular risk. Ang II plays a key role in the pathogenesis of CKD in essential hypertension. In particular, excess of Ang II stimulation induces endothelial dysfunction, which, in the kidneys, can evolve in glomerulosclerosis, tubulointerstitial fibrosis and vascular sclerosis. In the absence of pharmacological intervention, these abnormalities are responsible of development of overt nephropathy that culminates in end-stage of renal disease (ESRD). Clinical manifestations of hypertension-induced nephropathy are: presence of macroalbuminuria or proteinuria, decrease of glomerular filtration rate (GFR), increase in serum creatinine levels. Although achievement of a tight control of BP is important goal to prevent CKD, this strategy, alone, often is not enough to prevent the development and progression of CKD. The benefit of ACE inhibitor therapy in reducing proteinuria and the progression of CKD in non-diabetic patients are known since 1990s; similarly, beneficial effects has been demonstrated also for ARBs in nondiabetic nephropathies [95]. Thus, antihypertensive drugs that interfere with RAS confer additional renoprotective benefits compared with other classes of antihypertensive agents. The first convincing demonstration of the ability of ACE-inhibitors to interfere with the progression of CKD comes from the REIN study. This study showed that although BP control did not differ between the two treatment groups, patients who had proteinuria of ≥3 g/day and were treated with the ACE inhibitor showed a significant lower rate of decline in GFR and a reduced risk for doubling serum creatinine or end-stage renal failure as compared with patients who received conventional therapy [28]. The favorable effects of ACE-inhibitors [38] in the delay of CKD have been confirmed by several meta-analysis [37, 48]. The beneficial effects of many ARBs have been well documented in diabetic nephropathy. In fact, clinical trials with irbesartan, losartan, telmisartan and valsartan have been conducted in patients with type 2 diabetes and CKD. On the contrary, there are few clinical evidence of the nefroprotective effects of ARBs in patients with non-diabetic nephropathy. In details, The Japanese Losartan Therapy Intended for the Global Renal Protection in HyperTensive Patients (JLIGHT) study examined the effect of losartan in comparison with amlodipine after 12 months of treatment. This study showed that although losartan and amlodipine had a comparable antihypertensive effect, losaran based treatment significantly reduced the severity of proteinuria [34]. In addition, the Angiotensin II Receptor Antagonist Micardis in Isolated Systolic hypertension (ARAMIS) study compared the antihypertensive efficacy after 6 weeks of once-daily fixed doses of telmisartan 20, 40 or 80 mg versus hydrochlorothiazide 12.5 mg or placebo in patients (n = 1,039, aged 35–84 years) with isolated systolic hypertension. This study showed that, despite comparable reductions in systolic blood pressure with both drugs, telmisartan treatment significantly reduced urinary albumin excretion than hydrochlorothiazide [102].
Experimental evidence: In last years is has been demonstrated that CKD is characterized by of chronic inflammation associated with oxidative stress, endothelial dysfunction, and vascular calcification. Moreover, it has also been documented that Ang II regulates not only the hydro-saline homeostasis and peripheral vascular resistances but, exerts also a control on cell growth, inflammation, and fibrosis [75]. Together these experimental evidence indicate that Ang II plays a pivotal role in the genesis of CKD in essential hypertension by modulating the redox status and the immune system. In fact, Ang II increases tumor necrosis factor-production in the kidney, as well as, upregulates other proinflammatory mediators, including interleukin 6, monocyte chemotactic protein-1, and nuclear factor-B [94], resulting in a variety of glomerular insults. These results allow the hypothesize that Ang II is involved into the pathogenesis of CKD by modulating the activation and infiltration of immunocompetent cells There are several evidence showing that some of the beneficial effects of the RAS blockade may be related to anti-inflammatory properties of ACE-inhibitors and ARBs [91]. In particular, it has been reported in monocytes that exposure to captopril affects the cytokine-induced translocation of nuclear factor-kB translocation from the cytoplasm to the nucleus [3]. Furthermore, it has been reported in patients with ESRD, that ACE-inhibitor-based treatment reduces plasma levels of plasma levels of tumor necrosis factor-α and C-reactive protein [90].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


