(1)
University of Ottawa The Ottawa Hospital, Ottawa, ON, Canada
Angiotensin-converting enzyme (ACE) inhibitors and angiotensin II receptor blockers (ARBs) play a pivotal role in the management of heart failure (HF) and hypertension. These agents are mildly cardioprotective and increase survival in patients with:
HF.
Left ventricular (LV) dysfunction.
Acute myocardial infarction (MI). The Survival of Myocardial Infarction Long-Term Evaluation (SMILE) study (Ambrosioni et al. 1995) showed that zofenopril administered to patients with acute anterior infarction improved survival.
Hypertension with LV hypertrophy (LVH).
Hypertension with diabetes and proteinuria.
Patients at high risk for cardiovascular events, as indicated by the Heart Outcomes Prevention Evaluation (HOPE) study (HOPE Investigators 2000). But in this hallmark RCT, unfortunately, optimal therapy (beta-blockers, aspirin, and statins) was administered to too few patients. One may question whether the benefits associated with ramipril in this study would have been maintained if patients had been treated with appropriate regimens of aspirin, beta-blockers, and lipid-lowering agents (Weinsaft et al. 2000 ). (See HOPE study under ramipril.)
Cardioprotection appears to be exaggerated and many people at low risk are prescribed these agents, in particular ARBs, which are not adequately proven to save lives or reduce stroke rates significantly; see discussion under TRANSCEND and PRoFESS (2008) trials.
Tissue angiotensin II production appears to be an important modulator of tissue function and structure. Angiotensin II produced in cardiac myocytes has been shown to play a role in stretch-induced hypertrophy and in the process of myocardial remodeling post infarction (Pfeffer and Braunwald 1990 ).
Three classes of ACE inhibitors have been developed. Most ACE inhibitors except captopril and lisinopril possess a carboxylic radical, are transformed in the liver to the active agent, and are thus prodrugs.
Class I: Captopril is not a prodrug; it is the active drug, but with metabolism, the metabolites are also active. Only captopril and zofenopril contain a sulfhydryl (SH) group.
Class II: All other available agents except lisinopril are prodrugs and become active only after hepatic metabolism to the diacid (Table 3-1).
Table 3-1
Pharmacologic profile and dosages of ACE inhibitors
Benazepril
Captopril
Cilazapril
Enalapril
Fosinopril
Lisinopril
Perindopril
Quinapril
Ramipril
Trandolapril
USA + Canada
Lotensin
Capoten
Inhibace
Vasotec
Monopril
Prinivil
Aceon Zestril
Accupril
Altace
Mavik
UK
–
Capoten
Vascace
Innovace
Staril
Carace, Zestril
Aceon
Accuprin
Tritace
Gopten/Odrik
Europe
Cibace
Lopril, Lopirin
Inibace
Xanef, Renitec
Carace, Zestril
Acertil
Accupro
Tritace
Gopten
Prodrug
Yes
No
Yes
Yes
Yes
No
Yes
Yes
Partial
Yes
Action
Apparent (h)
1
0.5
2–4
2–4
3–6
Peak effect (h)
2
1–2
4
4–8
3–6
Duration (h)
12–24
8–12
>24
12–24
24–30
24–48
Half-life (h)
10–11
2–3
>40
11
>24
13
>24
>24
14–30
24
Metabolism
–
Partly hepatic
Hepatic
None
Partial
Elimination
Renal
Renal
Renal
Renal
Renal + heptatic
Renal
Renal
Renal
Renal
Renal
SH group
No
Yes
No
No
No
No
No
No
No
No
Tissue specificity
No
No
Yes
No
Yes
No
Yes
Yes
Yes
Equivalent dose
10 mg
100 mg
2.5
20 mg
10
20 mg
3
15
10 mg
2
Initial dose
5–10 mg
6.25 mg
1.5
2.5 mg
5
2.5 mg
2
2.5–5
2.5–5
0.5
Total daily dose
Hypertension
10–20 mg
25–150 mg
1.5–5 mg
5–40 mg
5–40 mg
5–40 mg
2–8 mg
5–40 mg
2.5–15 mg
1–4 mg
Heart failure
–
75–150 mg
–
10–35 mg
–
10–35 mg
–
Dose frequencya
1 daily
2–3 daily
1 daily
1–2 daily
1 daily
1 daily
1 daily
1 daily
1 daily
1 daily
Supplied, tabs
5, 10, 20, 40 mg
12.5, 25, 50, 100 mg
1,2.5, 5 mg
2.5, 5, 10, 20 mg
10, 20 mg
2.5, 5, 10, 20, 40 mg
2, 4 mg
5, 10, 20, 40 mg
1.25, 2.5, 5, 10 mg
0.5, 1, 2 mg
Class III: Lisinopril is not a prodrug and is the only water-soluble agent; it is excreted unchanged by the kidneys. Lipid solubility does not confer clinical benefits beyond those observed with lisinopril.
The pharmacologic features and dosages of ACE inhibitors are given in Tables 3-1 and 3-2 and in Chap. 8.
Table 3-2
Profile of angiotensin II receptor blockers
Drug | Active metabolite | Bioavailability (%) | Half-life (h) | Food effect | Dose once daily (mg) |
|---|---|---|---|---|---|
Candesartan | No | 15 | 9 | None | 4–16 |
Irbesartan | No | 60–80 | 11–15 | None | 150–300 |
Losartan | Yes | 33 | 6–9 | Minimal | 50–100 |
Valsartan | No | 25 | 6 | 50 % | 80–160 |
Mechanism of Action
Vascular stretch of the renal afferent arteriole and the sodium concentration in the distal tubule, sensed by the macula densa and an interplay of beta-adrenergic receptors, control the release of renin from the juxtaglomerular cells located in the media of the afferent renal arteriole (Davis and Freeman 1976; Torretti 1982; Reid 1985).
Stimuli to the release of renin include:
A decrease in renal blood flow (ischemia), hypotension, and reduction of intravascular volume
Sodium depletion or sodium diuresis
Beta-adrenoceptor activation
The enzyme renin is a protease that cleaves the leucine 10–valine 11 bond from angiotensinogen to form the decapeptide angiotensin I (Ganong 1984). ACE now cleaves histidine–leucine from angiotensin I, resulting in the formation of angiotensin II, which causes:
Vasoconstriction about 40 times more intense than that caused by norepinephrine. Vasoconstriction occurs predominantly in arterioles and, to a lesser degree, in veins; this action is more pronounced in the skin and kidney, with some sparing of vessels in the brain and muscle.
Renal effects: marked sodium reabsorption occurs in the proximal tubule.
Adrenal effects: aldosterone release enhances sodium and water reabsorption and potassium excretion in the renal tubule distal to the macula densa. Angiotensin II also promotes release of catecholamines from the adrenal medulla.
Increased sympathetic outflow and facilitated ganglionic stimulation of the sympathetic nervous system (Ganong 1984; Munzel and Keaney 2001 ).
Modest vagal inhibition, which may explain the lack of tachycardia in response to the marked vasodilator effect of ACE inhibitors.
Enhanced antidiuretic hormone secretion, resulting in free water gain.
ACE inhibitors are competitive inhibitors of angiotensin-converting enzyme, and therefore they prevent the conversion of angiotensin I to angiotensin II (see Fig. 3-1). The consequences of this action are as follows.
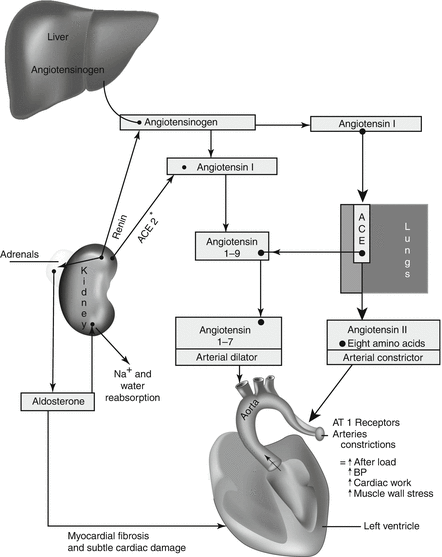
Fig. 3-1.
Renin–angiotensin–aldosterone system: action on the heart and arterial system. Asterisk: ACE 2 may counteract some of the effects of ACE activity. Angiotensin II action, however, must prevail to maintain adequate blood pressure to the brain and vital organs during catastrophic events: nature’s way of survival; ACE, angiotensin-converting enzyme. Angiotensin II activates two subtypes of angiotensin II receptors: AT1 and AT2, but only AT1 mediates clinical effects of angiotensin. Reproduced with permission from Khan MG, Encyclopedia of Heart Diseases, 2nd edition. New York: Springer Science + Business Media; 2011, p. 2. With kind permission from Springer Science + Business Media.
Arteriolar dilation causes a fall in total systemic vascular resistance, blood pressure, and afterload; these three terms are interrelated but are not synonymous (Burnier 2001).
Sympathetic activity decreases because of attenuation of angiotensin-related potentiation of sympathetic activity and release of norepinephrine. The diminished sympathetic activity causes further vasodilation with additional reduction in afterload and some decrease in preload. It is because of this further indirect antisympathetic and vagal effect that heart rate is not increased by ACE inhibitors, as opposed to several other groups of vasodilators.
Reduction in aldosterone secretion promotes sodium excretion and potassium retention.
Vascular oxidative stress is favorably influenced (Munzel and Keaney 2001) because vascular superoxide is reduced. Thus, ACE inhibitors are believed to have important antioxidant properties superior to those of vitamin E and other antioxidants. A review by Burnier (2001) gives details and relevant references. Vascular wall endothelium, smooth muscle, and fibroblasts contain enzyme systems that use nicotinamide adenine dinucleotide and its reduced form (NADH and NADPH) for the production of superoxide anion that is increased in response to angiotensin II. ACE activity has been noted to increase in atheromatous plaques, and inhibition appears to influence inflammatory reaction favorably within the arterial wall. Angiotensin II is a mitogen for vascular smooth muscle cells that can be inhibited. Superoxide is a major source of hydrogen peroxide; thus, smooth muscle cell proliferation may be limited. Also, nitric oxide (NO) activity appears to improve because superoxide reacts with NO (Burnier 2001).
Increased free water loss is caused by blocking of angiotensin-mediated vasopressin release, resulting in some protection from dilutional hyponatremia. This action is important in patients with severe HF.
Increased bradykinin-converting enzyme is the same as kinase II, which causes degradation of bradykinin. The accumulation of bradykinin stimulates release of vasodilatory NO and prostacyclin that may protect the endothelium and contribute to arterial dilation and to a decrease in peripheral vascular resistance. Thus, indomethacin and other prostaglandin inhibitors reduce the effectiveness of ACE inhibitors. Captopril has been shown to be uricosuric (Leary and Reyes 1987), and it reduces hyperuricemia.
Arteriolar hyperplasia is decreased. ACE inhibitors have been shown to decrease arteriolar hyperplasia caused by hypertension. Therapy with cilazapril for 1 year appears to correct the structural and functional abnormalities in the resistance arteries of patients with mild essential hypertension.
ACE gene polymorphism contributes to the modulation and adequacy of the neurohormonal response to ACE inhibitor long-term administration in HF (Cicoira et al. 2001). Patients with HF with aldosterone escape have been shown to have a higher prevalence of DD genotype compared with patients with normal aldosterone levels (Cicoira et al. 2001). The antihypertensive response to ACE inhibition has also been shown in a small series to be more pronounced in subjects with ACE DD genotype than in those with the ACE-11 genotype (Cicoira et al. 2001). Genetic screening of large populations of patients, however, remains controversial.
ACE Inhibitors Versus Other Vasodilators
The reversal of iatrogenic hypokalemia caused by ACE inhibition is an important asset in the management of patients with hypertension and HF, who often require diuretic therapy.
The suppression of ADH activity by ACE inhibitors decreases free water gain, which is useful in the management of the hyponatremic patient with HF. This salutary effect is not observed with other vasodilators.
Both ACE inhibitors and calcium antagonists are effective in preventing LVH and also cause it to regress when present, but other vasodilators do not consistently prevent hypertrophy or cause regression. LVH is an independent risk factor for sudden death, and its prevention is therefore an important aspect of pharmacologic therapy. ACE inhibitors are generally well tolerated, with few adverse effects, whereas fewer than 33 % of patients tolerate hydralazine or alpha1-blockers after 6 months of therapy at doses sufficient to achieve goal blood pressure. ACE inhibitors cause marked arteriolar vasodilation and a significant decrease in venous tone, resulting in a decrease in afterload and preload. In contrast with other vasodilators, with the exception of calcium antagonists, they cause afterload reduction, but their administration sets in motion compensatory mechanisms that have several effects tending to counteract their beneficial action.
Prazosin and other alpha1-blockers cause a decrease in afterload and a mild decrease in preload, but they increase heart rate and cardiac ejection velocity, resulting in a deleterious rate of rise of aortic pressure. These agents cause sodium and water retention that necessitates an increase in prazosin dosage and often requires added diuretic therapy. Tachyphylaxis occurs, and clinical trials have proved prazosin to be ineffective for prolonging life in the setting of HF.
Hydralazine has had extensive clinical testing. The Veterans Administration Vasodilator Heart Failure Trial (V-HeFT II) (Cicoira 2001) showed the drug to be effective in HF when combined with the venodilator effect of nitrates. Hydralazine causes a marked enhancement in heart rate and cardiac ejection velocity. This action is undesirable in patients with ischemic heart disease and limits the usefulness of this agent. Other vasodilators of this class, including alpha1-adrenergic receptor blockers (trimazosin, indoramin, terazosin), cause undesirable effects similar to those of prazosin and hydralazine. Other vasodilators, except for those that have a primary renal and adrenal action, of necessity cause a stimulation of the renin–angiotensin system as well as an adrenal release of catecholamine and sympathetic stimulation to compensate for the arteriolar dilation. These untoward effects, however, allow for the occasional combination of one of the aforementioned vasodilators with an ACE inhibitor.
Nitroglycerin is predominantly a venous dilator and decreases preload. A minimal decrease in afterload occurs with the use of intravenous (IV) nitroglycerin but not oral nitrates. These agents are useful in the management of chronic HF only when they are added to arteriolar vasodilators.
Clinical Indications
Hypertension
ACE inhibitors and selected ARBs are indicated for hypertension of all grades (telmisartan and olmesartan are not recommended). Some would argue that there are many ACE inhibitors and ARBs that have not been associated with a signal of increased cardiovascular death, so why not prescribe one of those agents (Ingelfinger 2011)? Their low side effect profile, especially with the quality-of-life advantages over some other antihypertensive agents, has resulted in their inappropriately widespread use.
But compared with calcium antagonists, they are surprisingly weak antihypertensive agents; they achieve reasonable reduction in blood pressure in <45 % of individuals and reach ~66 % only when a diuretic is added.
As outlined earlier, their built-in protection from reflex sympathetic stimulation, resulting in an increase in heart rate and the rate of rise of aortic pressure, is a major advantage over alpha1-adrenergic receptor antagonists (alpha-blockers) and similar vasodilators. ACE inhibitors and ARBs retain potassium and avoid the need for gastric-irritating potassium supplements.
Rebound hypertension observed after withdrawal of clonidine, guanabenz, guanfacine, methyldopa, and, rarely, calcium antagonists and beta-blockers is not a feature of ACE inhibition.
ACE inhibitors are most effective in young patients, aged less than 55 years, with essential hypertension, who usually have increased renin activity. In this subset of patients, ACE inhibitors or ARBs prescribed as monotherapy are effective in about 45 % of cases. In patients with more severe hypertension, ACE inhibitors in combination with diuretics are effective in up to 70 %. ACE inhibitors are slightly less effective in reducing blood pressure in nonwhite patients and in the elderly, although studies indicate a sufficiently good response to justify a trial of ACE inhibitors or ARBs as monotherapy in the elderly when other agents are contraindicated or poorly tolerated. The antihypertensive action of ACE inhibitors is multifactorial and partially depends on the renin and sodium status. Thus, it is not surprising that ACE inhibitors have been shown to be effective in elderly patients with low renin status.
ACE inhibitors and ARBs are particularly effective in lowering blood pressure in patients with high renin–angiotensin states, such as:
In combination with moderate to high doses of diuretics and calcium antagonists for the management of resistant hypertension.
In malignant hypertension.
In hypertension resulting from oral contraceptive use.
In coarctation of the aorta.
Immediately after dialysis in patients with chronic renal failure, when sodium and volume depletion is associated with enhancement of the renin–angiotensin system and responds to ACE inhibition (Weber 1999).
For the management of hypertensive patients with concomitant HF, for which ACE inhibitors are ideal. In this subset of patients, the systolic blood pressure should be maintained at less than 140 mmHg. The use of ACE inhibitors complements therapy with diuretics because diuretic use in this context stimulates the renin–angiotensin system.
ACE inhibitors are highly recommended in the following clinical situations:
In the hypertensive diabetic patient, ACE inhibitors are first-choice agents because they do not adversely affect glucose metabolism, they have a proven effect in reducing diabetic proteinuria, and there is some evidence suggesting prolongation of nephron life. ACE inhibition enhances insulin-mediated uptake of glucose, and this effect may be important in the management of hypertensives with diabetes.
These drugs are useful in hypertensive patients with hyperlipidemia because these agents produce no change in lipid parameters. When needed, a combination with a beta-blocking drug may provide cardioprotection.
Tissue ACE inhibition allows for the use of ACE inhibitors to decrease blood pressure in patients who have undergone nephrectomy.
Although these agents are very effective in patients with renovascular hypertension, they must be used, if at all, with extreme caution because severe renal insufficiency may occur in patients with bilateral renal artery stenosis or stenosis in a solitary kidney. Because ACE inhibitors cause dilation of the efferent arteriole, they may precipitate renal failure or the loss of a kidney.
Clinical Trials
PROGRESS: The Perindopril Protection Against Recurrent Stroke Study was a large, well-run RCT of 6,105 individuals with hypertension and previous stroke or transient ischemic attack.
The ACE inhibitor perindopril attained blood pressure goal in only about 42 % of trial patients. The addition of a diuretic was required in the majority of patients. No discernible reduction in the primary outcome, reduction in the risk of stroke, was observed for perindopril monotherapy (PROGRESS Collaborative Group 2001).
ACCOMPLISH: In a large RCT, a benazepril–amlodipine combination was superior to the benazepril–hydrochlorothiazide combination in reducing cardiovascular events (Jamerson et al. for the ACCOMPLISH Trial Investigators 2008). This is not surprising because diuretics are weak antihypertensive agents and calcium antagonists are the most powerful agents currently available. (See Chap. 22 for details.)
The post hoc observation of an adverse effect on mortality and morbidity in the subgroup receiving valsartan, an ACE inhibitor, and a beta-blocker raises concern about the potential safety of this specific combination (Cohn and Tognoni 2001).
Heart Failure
ACE inhibitors have provided a major improvement in the management of HF, resulting in both amelioration of symptoms and an increase in survival when they are used in combination with diuretics and digoxin (Cohn et al. 1991). The fall in cardiac output with HF triggers a compensatory response that involves enhancement of the sympathetic nervous system and the renin–angiotensin–aldosterone system. As a result of these adjustments, systemic vascular resistance and afterload are increased inappropriately, with further deterioration in cardiac performance, and a vicious circle ensues (see Chap. 12). ACE inhibitors play a vital role in halting the counterproductive pathophysiologic events that tend to perpetuate, rather than correct, cardiac decompensation.
The beneficial effects of diuretics in the management of HF are limited by excessive stimulation of the renin–angiotensin system; the addition of ACE inhibitors results in further amelioration. This improvement is partly the result of reduced arterial and venous tone, but changes in fluid and electrolyte balance are also important.
Stimulation of the sympathetic and the renin–angiotensin–aldosterone system causes intense sodium and water retention in the proximal and distal nephron. Also, an increase in venous tone occurs. Both adjustments result in an increase in filling pressure, which enhances preload. ACE inhibitors partially inhibit sodium and water retention and decrease venous tone, which produces a decrease in preload, an improvement or decrease in symptoms and signs of pulmonary congestion, and an increase in exercise tolerance. The improvement in functional capacity is superior to that observed with hydralazine and is similar to the combination of hydralazine and isosorbide dinitrate.
Clinical Trials
CONSENSUS: The Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS 1987) indicated an increased survival in New York Heart Association (NYHA) class IV patients treated for over 6 months with enalapril added to diuretics and digoxin. The 6-month mortality rate was 26 % in patients treated with enalapril, versus 44 % in those given diuretic and digoxin alone (P < 0.001). Forty-two percent of the group treated with enalapril showed functional class improvement compared with 22 % in the control group (P = 0.001).
SOLVD Investigators (1991): Patients with chronic heart failure and EF < or equal to 0.35 (New York Heart Association functional classes II and III), receiving conventional treatment for heart failure, were randomly assigned to receive either placebo (n = 1,284) or enalapril (n = 1,285) at doses of 2.5–20 mg/day in a double-blind trial. Results were as follows:
At 41.4 months, there were 510 deaths in the placebo group (39.7%), as compared with 452 in the enalapril group (35.2%), a modest 11.4 % reduction (reduction in risk, 16%; P = 0.0036).
Fewer patients died or were hospitalized for worsening heart failure (736 in the placebo group and 613 in the enalapril group; risk reduction, 26 % [95 % confidence interval, 18–34%; P < 0.0001]) (SOLVD Investigators 1991). But the exact reduction in hospital admissions for heart failure should be separated.
Although these agents have provided much relief for suffering patients and have significantly decreased the mortality, the effect is still modest and newer agents must be sought.
In Val-HeFT valsartan when combined with a beta-blocker caused significantly more cardiac events. A total of 5,010 patients with HF class II, III, or IV were randomly assigned to receive 160 mg of valsartan or placebo twice daily. There was no reduction in all-cause mortality. The incidence of the combined end point, however, was a modest 13.2 % lower with valsartan than with placebo P = 0.009), predominantly because of a lower number of patients hospitalized for heart failure: 455 (18.2%) in the placebo group and 346 (13.8%) in the valsartan group (P < 0.001). “Overall mortality was not reduced by valsartan administration” (Cohn and Tognoni 2001).
Unfortunately, less than 36 % of subjects received a beta-blocker and ~68 % received digoxin.
The post hoc observation of an adverse effect on mortality and morbidity in the subgroup receiving valsartan, an ACE inhibitor, and a beta-blocker raises concern about the potential safety of this specific combination (Cohn and Tognoni 2001).
V-HeFT 11: The V-HeFT II trial, at an average 2.5-year follow-up, showed a modest improvement in survival, the mortality rate being 33 % for enalapril added to diuretics and digoxin versus 38 % in patients given a diuretic and digoxin along with hydralazine and isosorbide dinitrate (Cicoira 2001).
It is clear that ACE inhibitors improve survival in certain categories of patients, and benefit is beyond question for patients with NYHA class IV HF (15). There is as yet no convincing evidence that these drugs decrease mortality in patients with NYHA class II HF, and although V-HeFT II results suggest some benefit in class II patients, this is not statistically significant (see Chap. 12).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


