Figure 1-1.
Anatomy and distribution of the cardiac nervous systems. The cardiac sympathetic nerve ends (blue) are projected from sympathetic neurons in the stellate ganglia, which are bilateral to the thoracic vertebra. The stellate ganglia originate from the spinal cord (dorsal root ganglia or preganglia). The cardiac parasympathetic nerves (red) extend from parasympathetic neurons in the cardiac ganglia, which are located at the base of both atria, and are direct branches of the vagus nerve in the human from the brainstem (10th cranial verve). The sensory nerves (green) project to the upper thoracic dorsal horn via dorsal root ganglia, which are responsible for pain perception and initiation of a protective response during cardiac injury. The inset indicates the transverse section of the heart and demonstrates the distribution of sympathetic nerve fibers within the left ventricle. The sympathetic nerve fibers run in the epicardial area and then individually dive into the myocardial wall, and display an epicardial-to-endocardial gradient in the heart [2]. Maintenance of this physiologic innervation gradient is required for sustaining a physiologic repolarization gradient across the ventricular walls. Collapse of the innervation gradient may have an adverse effect on the electrical phenotype of the ventricular wall, resulting in arrhythmia (Data from Kimura et al. [12]).
Sympathetic Neuronal Synapse in the Heart
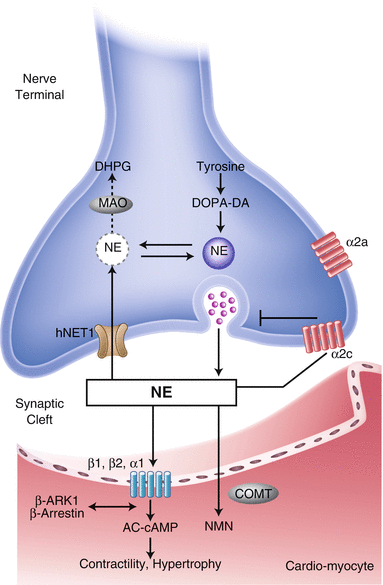
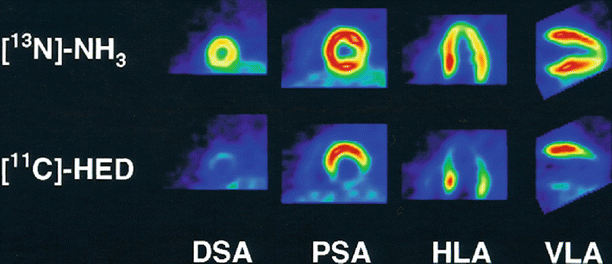
Figure 1-2.
Sympathetic neuronal synapse in the heart. The sympathetic neuronal transmitter norepinephrine (NE) is synthesized from tyrosine. First, it is converted to dopamine by cytoplasmic enzymes. Dopamine then is transported to intraneuronal vesicles, where NE is synthesized and stored. Upon neuronal stimulation, the vesicles fuse with the presynaptic membrane and release the NE into the cleft. The released NE then binds to the postsynaptic α- and β-adrenergic receptors, which are G-protein–coupled receptors, to transduce signals to improve excitation–contraction coupling of the myocardium and to increase heart rate and force. Cleft NE also activates the presynaptic α2-adrenergic receptors to inhibit further NE release. The effect of the NE is terminated by two mechanisms: approximately 80 % of the NE is taken up to the presynaptic area by an ATP-dependent active process via a transporter called human norepinephrine transporter 1 (hNET1; uptake-1 system), whereas the reminder spills over into the nonneuronal tissue and bloodstream (uptake-2 system). Thus, several molecules in this pathway may serve as targets for the functional imaging of sympathetic tone in the heart. For example, to target the neurotransmitter NE, 123I-mIBG and 11C-HED were developed. Both radiotracers mimic the neuronal transport and storage process of NE and represent cardiac sympathetic activity. hNET1 also may be an ideal target to represent the uptake-1 system activity in the heart. AC adenyl cyclase, AMP adenosine monophosphate, cAMP cyclic adenosine monophosphate, COMT catechol-O-methyltransferase, DA dopamine, DHPG dihydroxyphenylglycol, DOPA dihydroxyphenylalanine, MAO monoamine oxidase, NMN normetanephrine (Data from Haider et al. [13]).
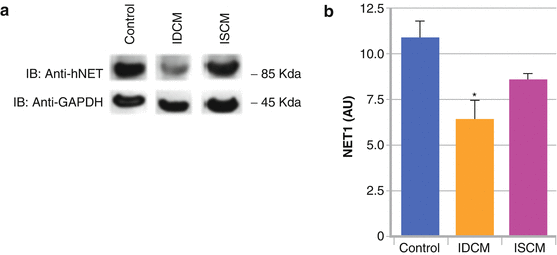
Figure 1-3.
Down-regulation of the presynaptic hNET1 in heart failure. In patients with heart failure (ischemic or nonischemic), initially there is increased sympathetic activity with increased release of NE from the presynaptic vesicles as a compensatory reaction to contribute to increased cardiac contractility. (a, b) There is corresponding down-regulation of the hNET1 transporter (decreased uptake-1 system) in idiopathic dilated cardiomyopathy (IDCM) and ischemic cardiomyopathy (ISCM) [13], with a subsequent increase in quantities of NE in the synaptic cleft. Gradually, the compensatory mechanism is overwhelmed, and a persistent NE excess in the cleft eventually results in desensitization of the postsynaptic adrenergic receptors [5], myocyte apoptosis/myocellular loss [14], worsening heart failure, increased susceptibility to arrhythmia, and sudden cardiac death [15]. A small proportion of this NE spills into the bloodstream (increased uptake-2 system, with increasing plasma NE levels). These compensatory changes can be imaged with the NE analogue 123I-mIBG as decreased retention of the radiotracer in the presynaptic area and increased washout. GAPDH glyceraldehyde-3-phosphate dehydrogenase, IB immunoblot. *P < 0.05: intensity of the hNET1 bands mean SEM of four IDCM, four ISCM, and eight control hearts. Values were corrected by the respective values of GAPDH for each sample (Data from Haider et al. [13]).
Signal Transduction of the Postsynaptic Cardiac Adrenergic Receptors
The human genome encodes nine different adrenoceptor genes. They are grouped into three G-protein–coupled receptor (GPCR) families with three members each, namely the α1-adrenoceptors (α1A, α1B, and α1D), α2-adrenoceptors (α2A, α2B, and α2C), and β-adrenoceptors (β1, β2, and β3) [16]. In the human heart, the β1-adrenoceptor subtype is predominant, representing 75–80 % of total adrenoceptor density. The β2 subtype also is present, representing 15–18 %, whereas β3-adrenoceptors comprise the remaining 2–3 % [17, 18]. The β1 subtype mediates an increase in cardiac contractility, whereas the β2 subtype is responsible for vasodilatation. These receptors are not just signal transducers but also components of a complex and highly regulated signaling machinery. Adrenoceptors consist of seven transmembrane (TM)-spanning α-helical domains, an extracellular region (N terminus, three extracellular loops), and an intracellular region (three intracellular loops, C terminus).
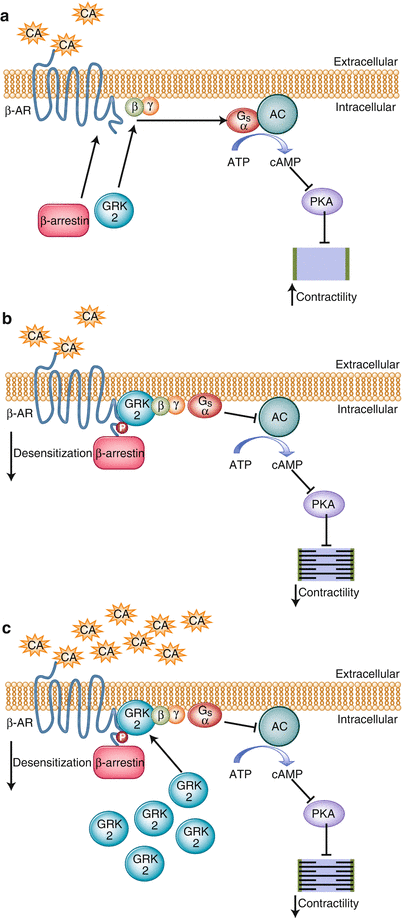
Figure 1-4.
Signal transduction of cardiac myocyte contraction and its regulation by cardiac adrenergic receptors. Under normal conditions (a), upon catecholamine binding, adrenoceptors change conformation, couple to and activate Gαs, and thereby initiate various intracellular signaling cascades that ultimately lead to increased contractility by activating adenylate cyclase (AC) and protein kinase A (PKA). β-adrenoceptor signaling is subject to agonist-promoted desensitization (b), a loss of function as a result of uncoupling from the signal transducing G protein, which occurs within a few seconds and diminishes receptor response to continuous or repeated agonist stimulation. At the molecular level, this rapid process can be induced by receptor phosphorylation by a family of kinases, termed G-protein-coupled receptor kinases (GRKs). The cytosolic GRK translocates and anchors to the membrane by bindingto Gβγ via its C terminus where it is able to interact with the agonist-occupied receptor and phosphorylate it, which begins the G protein uncoupling process. G protein activation is blocked by recruitment of β-arrestin molecules to the phosphorylated receptor. The β-arrestins then uncouple the receptor from its cognate G proteins, sterically hinder its further binding to them (functional desensitization), and subsequently target the receptor for internalization [19, 20]. In addition, receptors can be downregulated, a mechanism requiring several hours, by decreasing receptor synthesis or increasing receptor degradation or sequestration. In addition, β-adrenoceptors mRNA destabilization in response to agonist exposure may also occur [21]. After cardiac injury (c), GRKs levels are increased because of enhanced β adrenoceptor activation after stress-induced increases in catecholamines. The increase in GRKs help to prevent overstimulation of the β adrenoceptors initially, but over time a vicious cycle of chronically increased β adrenoceptor desensitization occurs and β1 downregulation [22], which lead to progressive worsening of the cardiac adrenergic and inotropic reserves, the hallmark of heart failure [23] (Data from Woodall et al. [24]).
Heterogeneity of Innervation in the Normal Heart
Clinical studies using 123I-mIBG scintigraphy have demonstrated heterogeneous sympathetic distribution in normal hearts, particularly in the inferior region. Such decreased inferior mIBG activity was found to be more prevalent in older subjects and more evident in men than women [1]. The findings were confirmed by others who showed that in men, mIBG uptake in the inferior wall was inversely correlated with age. There was a 16 % reduction in mIBG uptake in the 80-year-old age group compared with the 40-year-old age group [24]. Although the mechanism for this age-related decrease of mIBG in the inferior region of the left ventricular myocardium is not clear, it might be related to high parasympathetic activity in that area. In a study of mIBG uptake in high-level male athletes, the subset of athletes with sinus bradycardia (heart rate <50 bpm; mean age, 27 years) had significantly less inferior mIBG uptake (44 ± 13 %) than those with higher heart rates (heart rate >60 bpm; mean age, 34 years), who exhibited relatively preserved inferior mIBG uptake (72 ± 11 %, P < 0.01) [25]. These findings support the notion that high parasympathetic activity, as characterized by sinus bradycardia, may contribute to the inferior mIBG abnormalities, as do diaphragmatic attenuation artifacts.
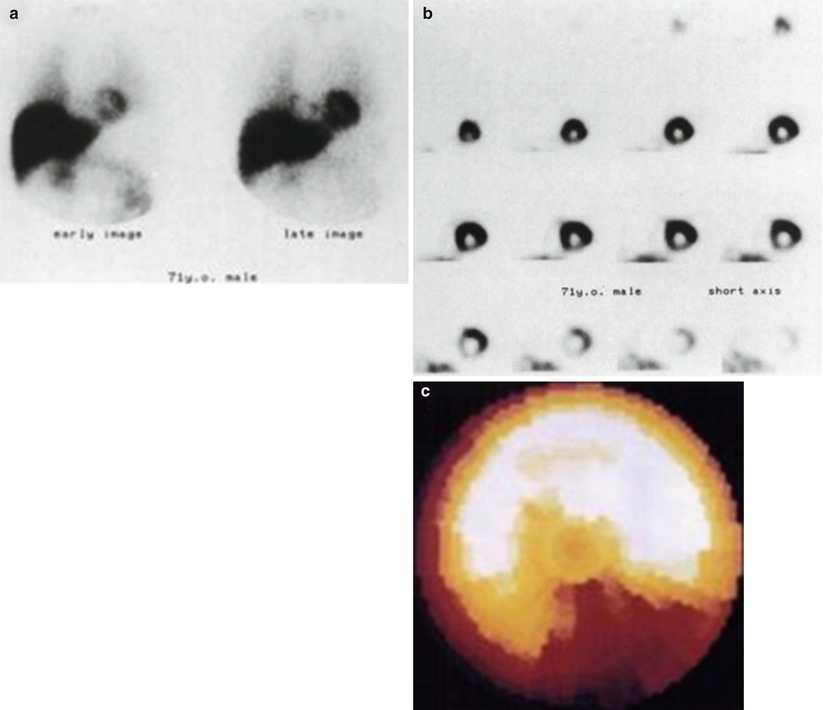
Figure 1-5.
Heterogeneity of Innervation in the Normal Heart. The images show a 71-yr-old man with malignant lymphoma before adriamycin administration. High accumulation of tracer in the myocardium is shown on the early and the late planar images (a). On the short-axis SPECT images (b) and bull’s-eye polar map (c), however, decreased mIBG uptake in the inferior region is noted (This research was originally published in JNM. Tsuchimochi et al. [1]. Figure 1.2. © by the Society of Nuclear Medicine and Molecular Imaging, Inc.).
Reinnervation in the Transplanted Heart
The transplanted heart lacks both sympathetic and parasympathetic innervation because the nerves are dissected during surgery. The absence of direct nerve stimulation leads to an inadequate balance between the sympathetic and parasympathetic systems. In the absence of parasympathetic stimulation, heart rate and myocardial contractility are both increased in the transplanted heart [26, 27]. On the other hand, the lack of sympathetic denervation and norepinephrine release prevents the heart rate from increasing enough to meet stress or exercise demands (chronotropic incompetency). The denervated heart becomes dependent on circulating norepinephrine for increasing the heart rate, contractility, and conduction via β1-receptor stimulation [28], which is minimal and delayed. Both parasympathetic [29] and sympathetic [30] reinnervation has been observed in transplanted hearts. Although the time course of reinnervation and the extent of reinnervation in the transplanted heart may vary, clinical studies have reported partial cardiac reinnervation between 1 and 15 years after cardiac transplantation [31–33]. Reinnervation may occur earlier in younger donor hearts and with a shorter surgical transplantation (ischemic) time. Reinnervation of the transplanted heart results in subsequent improvement in both left ventricular function and exercise tolerance in recipients [30].
Through the use of 123I-mIBG or 11C-HED PET imaging, studies have demonstrated partial reinnervation after transplantation [34–36]. Reinnervation has been found primarily in the basal anteroseptal region and, to a lesser degree, in the inferobasal and apical regions [34, 37]. The midventricular region of the myocardium typically demonstrates less reinnervation than the basal region. The findings of heterogeneous reinnervation may be explained partly by the fact that sympathetic neurons travel along arterial structures; thus, the basal regions are reached first by the newly grown fibers.
Molecular Regulation of Cardiac Nerve Development
Molecular signaling and regulation of cardiac autonomic development and innervation are not well elucidated yet. Understanding the mechanisms involved in neuronal development and reinnervation after injury is important in managing the diseased heart, especially with regard to conditions such as life-threatening ventricular arrhythmias after infarction, which may be related to sympathetic nerve sprouting. Studies have shown that cardiac autonomic neurogenesis and function are regulated tightly by a plethora of neurotrophic factors, which are divided into two classifications: the neurotrophin and glial cell line–derived neurotrophic factor (GDNF) families. The neurotrophin family is composed of several factors, including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophins 3 and 4/5 (NT3 and NT4/5), acting through the high-affinity tyrosine kinase receptors, tropomyosin receptor kinase (Trk), and lower-affinity neurotrophin receptor p75 (p75NTR). The GDNF family includes GDNF, neurturin, and artemin, which bind to GDNF family receptor-α (GFRα) and RET [38–40].
NGF, the prototypic member of the neurotrophin family, acts primarily through TrkA, which promotes growth, survival, and maintenance of sympathetic nerves [41, 42]. The level of NGF expression in the heart correlates approximately to the sympathetic innervation density. Overexpression of NGF in the mouse heart results in hyperinnervation and hyperplasia in stellate ganglia neurons [43]. On the other hand, sympathetic ganglion volume is reduced by 80 % at postnatal day 3 in mice with a disruption of the NGF gene [41]. In mice lacking the NGF receptor TrkA, no neurons remain at postnatal day 9 [39, 44]. NGF mediates axonal growth and synapse formation during development [45]. It is required for complete peripheral innervation of sympathetic ganglia targets [46] and is necessary and sufficient for synapse formation within sympathetic ganglia [47].
Different from neurotrophic factors such as NGF, as mentioned above, which act as chemoattractants in cardiac nerve development, some factors work as chemorepellents that inhibit nerve growth and distribution during development. Studies have shown that Sema3a, a member of the semaphorin family, is such a chemorepellent, affecting sympathetic neural patterning in the developing heart [47]. Sema3a is expressed in the subendocardium but not in the subepicardium, in contrast to the epicardial-to-endocardial gradient of sympathetic innervation (Fig. 1.6). Cardiac-specific Sema3a-overexpression mice had reduced sympathetic innervation and attenuation of the epicardial-to-endocardial innervation gradient. These results indicate that cardiomyocyte-derived Sema3a plays a critical role in cardiac sympathetic innervation patterning by inhibiting neural growth.
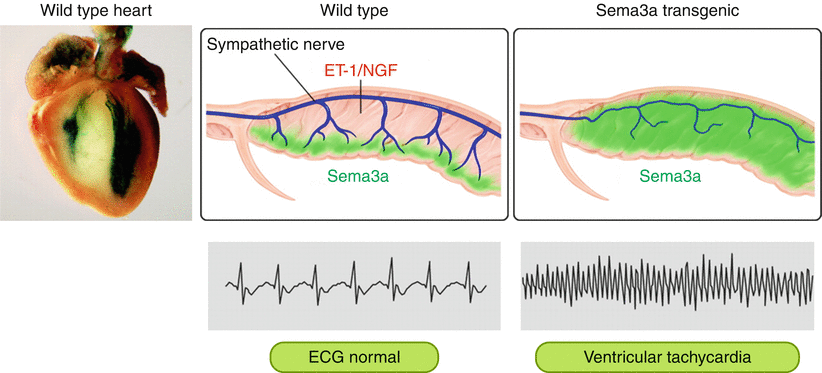
Figure 1-6.
Regulation of cardiac innervation patterning and maintenance of an arrhythmia free heart. Left, X-gal staining (green) of Sema3alacZ hearts at E15 demonstrates strong Sema3a expression in the subendocardium. Middle, Cardiac sympathetic innervation shows an epicardial-to-endocardial transmural gradient. This patterning is established by the balance between ET-1/NGF and Sema3a expression in the heart. Note that NGF is expressed abundantly in the working myocardium, whereas Sema3a is expressed specifically in the subendocardium. Right, Appropriate Sema3a mediated sympathetic innervation patterning is critical for the maintenance of an arrhythmia-free heart. Sema3a Transgenic mice display disruption of innervation patterning and are highly susceptible to ventricular tachyarrhythmias (From Kimura et al. [12]; with permission).
The GDNF family also is involved in sympathetic nerve regeneration in addition to its effect on parasympathetic nerve development [48]. Artemin, a neurotrophic factor of the GDNF family, was shown to promote the development of sympathetic innervation along blood vessels by enhancing sympathetic axon growth [49, 50]. Cocultures of sympathetic ganglionic neurons and cardiomyocytes demonstrated that GDNF promotes sympathetic axon growth toward cardiomyocytes more potently than NGF, with morphologic and functional synapse formation with the concomitant increase of β1-adrenergic receptors in cardiomyocytes [51, 52]. Under pathophysiologic conditions, the effect of GDNF was more potent than that of neurotrophic factors such as NGF, BDNF, or ciliary neurotrophic factor [53].
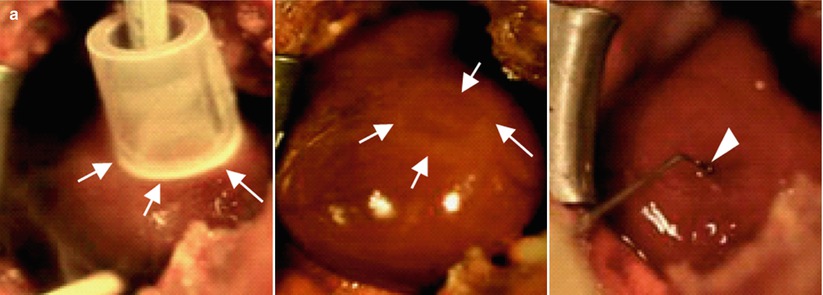
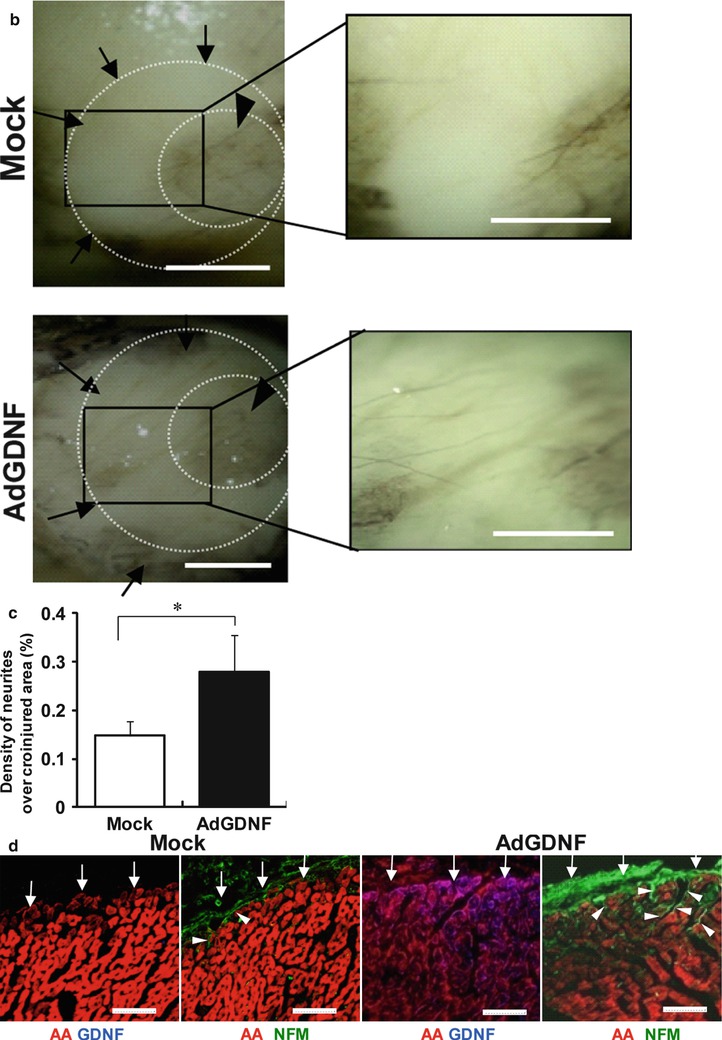
Figure 1-7.
Re-innervation in a rat model of denervation by cryoinjury. (a) Induction of denervation in rat hearts in vivo. A ring-shaped denervation was induced by cryoinjury on the epicardial surface of the left ventricular free wall (arrows in left and middle panels). Ad-GDNF was injected into the inner area of the injured ring (arrowhead in right). Ad-GFP was injected in controls. (b) Representative pictures of axon growth in the ring-shaped denervated area 5 days after the creation of cryoinjury. The hearts were isolated and whole-mount labeled for neurofilament-M (NFM, DAB stain). In the control heart (top), axons were detected only sparsely in the denervated area (arrows). In the Ad-GDNF-injected heart (bottom), in contrast, axons grew abundantly across the denervated area (arrows). Arrowheads indicate the uninjured central area (inner ring). Right panels show magnified images of the rectangular zones (injured areas). Bars indicate 1 mm. (c) The percentage of the denervated area covered by axons (NFMpositive). Values are means ± SD. *p < 0.05 vs control (n = 5). (d) Representative immunolabeling images of NFM (green), α-actinine (AA, red) and GDNF (blue) in the sections across the long axis of the rat hearts (denervated area). Note that GDNF was abundantly expressed in the myocardium, and the NFM-positive axons grew from the epicardial surface (arrows) into the mid myocardium in the AdGDNF heart (bottom). In the control heart (top), in contrast, GDNF was undetectable in the myocardium and the axons were recognized only scarcely on the epicardial surface. Bars indicate 50 mm (From Miwa et al. [51]).
Sympathetic Dysfunction in Cardiac Diseases
Compared with the normal heart, heterogeneity of sympathetic innervation in the diseased heart is more important clinically because it may cause ventricular arrhythmias and even sudden cardiac death. Imaging with 123I-mIBG scintigraphy or 11C-HED PET repeatedly has shown heterogeneous sympathetic innervation in diseased hearts. For example, after myocardial infarction, cardiac nerves undergo degenerative changes that result in heterogeneous innervations. The unbalanced sympathetic nerve innervation density may cause lethal arrhythmia [54].
Heart Failure
In the failing heart, cardiac sympathetic activity is increased, leading to pathophysiologic cardiac damage, depressed cardiac performance, and fatal arrhythmia. Although the underlying mechanisms are not fully understood, the phenomenon of increased innervation in the failing heart is associated with observations of decreased reuptake of norepinephrine into the presynaptic area (uptake-1 system) [55] and increased norepinephrine turnover or spillover in the synaptic cleft (uptake-2 system) [56]. This usually is reflected as decreased cardiac 123I-mIBG uptake on imaging. It should be mentioned that decreased cardiac activity on 123I-mIBG or 11C-HED PET often is described inappropriately as cardiac denervation in the literature. A true denervation usually is the result of nerve death in a totally infarcted area with complete absence of 123I-mIBG or 11C-HED PET activity.
Studies repeatedly have shown the utility of 123I-mIBG cardiac scanning in identifying heart failure patients with an increased risk of adverse cardiac events. A meta-analysis of 1755 patients from 18 studies confirmed that patients with a reduced heart-to-mediastinal (H/M) ratio have a poorer prognosis than those with a normal H/M ratio [8]. The ADMIRE-HF (AdreView Myocardial Imaging for Risk Evaluation in Heart Failure) study validated the independent prognostic value of the H/M ratio in patients with class II and III heart failure with left ventricular dysfunction [9]. The results showed that for patients with an H/M ratio less than 1.6, 2-year cardiac death and all-cause mortality were 11.2 % and 16.1 %, respectively, versus 1.8 % and 3.0 % for those with an H/M ratio ≥1.6. There was a progressive decline in both cardiac and all-cause mortality from greater than 20 % for H/M less than 1.10–0 % for H/M ≥1.8.
Cardiomyopathy
Dilated cardiomyopathy frequently is associated with regional neuronal dysfunction rather than a perfusion abnormality [57]. In patients with dilated cardiomyopathy, the inferobasal region is affected more commonly by neuronal dysfunction [58]. Diabetic cardiomyopathy often is associated with autonomic neuropathy. Imaging studies demonstrated a reduction in tracer uptake in the inferior wall of the left ventricle gradually spreading to adjacent segments. The extent of the reduced uptake was related to the severity of autonomic dysfunction as measured by conventional markers [59, 60]. Figure 1.8 shows an 11C-HED PET image from a patient with diabetes.