Fig. 10.1
AMPK structure. (a) AMPK domain structure of the three AMPK subunits (SID, subunit interaction domains; AID, autoinhibitory domain; CBM, carbohydrate-binding module; CBS, cystathionine-β synthase). (b) AMPK complex topology. Subunit interactions, secondary modifications (phosphorylations; myristoylation), and allosteric interactors (AMP; Act, putative activator at α/β interface). (c) Molecular structure and activation of the full-length AMPK heterotrimer (PDB 1CFF; Xiao et al. 2013). Binding of activating AMPK ligands AMP (γ-subunit) and A-769662 (β-subunit) has to be transduced to the α-subunit kinase domain for activation (see arrows), involving conformational changes. AMPK subunits α (green), β (blue), and γ (magenta) with α-subunit kinase domain, activation loop (AL), and regulatory interacting motif (α-RIM; Chen et al. 2013) indicated (dark green) and β-subunit carbohydrate-binding module (CBM) labeled; sequences missing in the structure (dashed) include the α-autoinhibitory domain (AID). Activation-relevant phosphosites (α-T172 in the activation loop and β-S108 in the CBM; red brown), activating ligands [A-769662 (red) and AMP (orange/yellow)], and kinase inhibitor staurosporine in the active site (green) are shown in spacefill representation. For further details see text (Figure modified from Viollet et al. 2014)
α-Subunit
The α-subunit contains a typical Ser/Thr protein kinase catalytic domain in its N-terminal part, with features conserved throughout the entire protein kinase superfamily (Hanks et al. 1988). It harbors a typical activation loop carrying the critical Thr172 residue which is phosphorylated for activation by AMPK upstream kinases like liver kinase B1 (LKB1) or Ca2+-calmodulin-dependent protein kinase kinase beta (CamKKβ ; see Chap. 4) (Hawley et al. 2003, 2005; Woods et al. 2003, 2005). This phosphorylation is considered as essential for AMPK activity (Hawley et al. 1996), although this has been recently challenged (Scott et al. 2014). Thr172 phosphorylation is also often used as a readout for AMPK activity, although this may not always be correct (see Chap. 4). The C-terminal part of the α-subunit carries different and important functionalities. Immediately downstream of the catalytic domain follows the autoinhibitory domain (AID) (Crute et al. 1998), which when fused to the kinase domain reduces AMPK activity as compared to kinase domain alone (Chen et al. 2009; Pang et al. 2007). In the further C-terminal sequence follows the so-called linker peptide and finally the very C-terminal region (~150 amino acids) which is required for association with the β-subunit. The latter also contains a long Ser/Thr-rich loop (not resolved in X-ray structures), as well as a nuclear export sequence [known to be functional in α2 (Kazgan et al. 2010)].
Of particular importance is the linker peptide, since it wraps around the γ-subunit like in a close embrace. A part of this linker peptide, first identified as α-hook, closely contacts γ-subunit (Xiao et al. 2011). Chen et al. (2013) more recently corrected the amino acid register for the electron density in this region, revealing the true γ-interacting sequence termed α-regulatory subunit interacting motif (α-RIM), interacting with CBS site 3 (see below) and with a pocket formed by a newly observed β-subunit loop. More recent structures of the near full-length heterotrimers confirmed these interactions of α-linker with β- and γ-subunits (Xiao et al. 2013) and its role in moving AID away from the kinase domain in the activated state (Li et al. 2014). In addition to Thr172, the α-subunit can be phosphorylated on several other residues both in vitro and in vivo. Most of these phosphorylations occur in the Ser/Thr-rich loop and seem to inhibit the activating Thr172 phosphorylation (see Chap. 4). Further structural studies will be necessary to delineate covalent and non-covalent activation of the kinase domain in molecular detail.
β-Subunit
The regulatory β-subunit represents the core of the heterotrimeric complex, since it provides a scaffold for binding of catalytic α- and regulatory γ-subunits. The N-terminal domain of the β-subunit carries an additional regulatory element, the conserved carbohydrate-binding module (CBM; also called glycogen-binding domain, GBD). Its structure complexed to beta-cyclodextrin has been solved (Polekhina et al. 2005), and it was shown that glucose α1-6-branched glycogen can behave as allosteric inhibitor, negatively regulating AMPK phosphorylation by its upstream kinases (McBride et al. 2009). In addition, CBM may serve to recruit AMPK to glycogen-bound downstream targets such as glycogen synthase (Hardie and Sakamoto 2006). The recent near full-length AMPK structures confirmed that glycogen binding moves CBM away from the α-kinase domain, while binding of pharmacological activators 991 and A769662 and autophosphorylation of β-S108, both at the α/β-interface (see Chap. 4), closely attach CBM to the kinase domain (Xiao et al. 2013; Li et al. 2014). The former conformation seems to be rather inhibitory, while the latter strongly activates AMPK. Thus, CBM is part of an allosteric regulatory site, which may also sense cellular energy reserves in the form of glycogen and mediate effects of certain pharmacological activators. The large very N-terminal portion of the β-subunit is not resolved in the known X-ray structures, and its function is not entirely clear. An N-terminal myristoylation affects activation (Oakhill et al. 2010) and could mediate membrane interaction. The sequence could also be involved in the interaction of AMPK with other proteins (Klaus et al. 2013).
γ-Subunit
All γ-subunits contain in their C-terminal part four tandem cystathionine β-synthase (CBS) repeats, a motif named by analogy to the cystathionine β-synthase in which it was first identified (Bateman 1997). In AMPK γ-subunits, the four CBS sites (numbered CBS 1 to CBS 4 according to their occurrence in the sequence) constitute a flattened disk with one CBS repeat in each quadrant and two pairs of CBS motifs assembling into a so-called Bateman domain. Four potential binding sites for adenylates (AMP, ADP, ATP) are created in the cleft between the CBS motifs, numbered according to the CBS motif that provides the conserved Asp for adenine ribose interaction (Kemp 2004). According to several crystal structures of the mammalian γ1-subunit in the presence of various nucleotides (Chen et al. 2012, 2013; Xiao et al. 2007, 2011, 2013), it appears that only CBS sites 1, 3, and 4 are functional, while the site 2 is different and always empty. The γ2- and γ3-isoforms contain long N-terminal extensions, which can be subject to truncation by RNA splicing, and whose molecular structure and function are currently unknown. The different γ-subunit isoforms and splice variants may be involved in protein/protein interaction and confer different cellular localization and function (Pinter et al. 2012).
10.3 Localization
Tissue Specificity; Cardiac AMPK
AMPK isoforms show some differences in their tissue- and developmental-specific expression patterns, although the physiological significance is still uncertain. There is no strict tissue specificity of AMPK isoforms, but increasing evidence suggests that a given tissue expresses a specific subset of AMPK heterotrimers which may be linked to particular signaling pathways in this tissue (Table 10.1). Studies with transgenic mice lacking specific α- and β-subunits have contributed to progress in this field (Viollet et al. 2009). While the α1β1γ1 complex is probably the most abundant in a vast majority of cell types, differences seem to occur in the amount of additional isoforms in a given tissue.
Table 10.1
Tissue expression of AMPK subunit isoforms
Isoform | Heart | Skeletal muscle | Brain | Liver | Lung |
|---|---|---|---|---|---|
Alpha 1 | ++ | ++ | ++ | ++ | ++ |
Alpha 2 | + | + | + | ||
Beta 1 | ++ | + | ++ | ++ | + |
Beta 2 | ++ | ++ | ++ | + | |
Gamma 1 | ++ | + | ++ | + | + |
Gamma 2 | +a | + | + | ||
Gamma 3 | + |
The heart contains high levels of the α2-isoform, which is much less expressed in the skeletal muscle and liver and almost absent in the brain. The β2-isoform is abundant in the heart and also in the muscle and brain. In addition to the γ1-isoform, the heart expresses a specific intermediate-length γ2-splice variant (γ2-3B), while γ3 seems to be quite specifically expressed only in the skeletal muscle (Stapleton et al. 1996; Thornton et al. 1998; Pinter et al. 2012). There are also pathological and developmental changes in cardiac AMPK expression. The α2-, β2-, and γ2-isoforms are all upregulated by pressure overload or heart failure in rodents, although in patients rather the content of α1, β1, and γ2 (an intermediate form) increases with different forms of cardiomyopathy (Tian et al. 2001; Kim et al. 2012a). During embryonic development in rodents, γ1 increases, while high levels of γ3 disappear, and the embryonically predominant full-length γ2-form is replaced by γ2-3B in the heart but by short γ2b in other tissues (Pinter et al. 2012). These developmental and tissue particularities may also explain why γ2-gene mutations in the CBS domains cause hereditary hypertrophic cardiomyopathy but no other pathological symptoms (see Chap. 6). Full-length γ2 and γ2-3B share an N-terminal domain with unknown function that could localize the AMPK complex to specific compartments or signaling pathways (Pinter et al. 2012). Total cardiac AMPK activity increases after birth, contributing to the switch toward the predominant use of fatty acids (Makinde et al. 1997). AMPK levels may also be determined by ubiquitin-dependent protein degradation (Qi et al. 2008; Moreno et al. 2010).
Subcellular Distribution
The subcellular distribution and recruitment of AMPK isoforms to specific cellular sites is increasingly recognized as an important factor for their signaling function. AMPK is generally observed as a soluble complex with diffuse cytosolic localization. However, at least α2-containing complexes in their activated form, e.g., after exercise in the skeletal muscle, also translocate into the nucleus to phosphorylate nuclear substrates such as transcription factors, histones, and histone deacetylases (McGee et al. 2003, 2008; Suzuki et al. 2007). Minor portions of AMPK may associate with cellular structures like specific membranes, where processes are regulated by AMPK (e.g., ion channel activity, cell polarity, or cell junction formation) (Forcet and Billaud 2007; Andersen and Rasmussen 2012; Nakano and Takashima 2012; Ramírez Ríos et al. 2014). Myristoylation of the AMPK β-subunit can localize the kinase complex to membranes and increases its activability, thus possibly favoring activation of membrane-bound complexes (Suzuki et al. 2007; Oakhill et al. 2010).
Multiprotein Complexes
AMPK can also be recruited into specific complexes via interaction with its upstream kinases, downstream substrates, or more general with scaffolding proteins. However, the AMPK interactome is only partially known so far from some targeted and non-biased interaction studies conducted by us and others (e.g., Behrends et al. 2010; Klaus et al. 2012), and more research is needed on this issue, in particular in the heart. AMPK interaction with LKB1, its major upstream kinase in the heart, could recruit AMPK to places of LKB1 localization, including the mitochondrial surface or E-cadherin in adherens junctions (Sebbagh et al. 2009). Close co-localization of both, AMPK and LKB1, can also be mediated by membrane interaction of both, farnesylated LKB1 and myristoylated AMPK (Houde et al. 2014).
Scaffolding proteins can in principle provide high specificity in cell signaling by isolating activated kinases from bulk signaling and directing the information flow into specific pathways. In the heart, for example, AMPK competes with p38 MAPK for binding to the scaffolding protein TAK-1–binding protein-1, thus blunting p38 activation during ischemia (Li et al. 2005). Mitochondrial VDAC may represent yet another anchoring protein that recruits AMPK to this organelle (Strogolova et al. 2012). Most interestingly, the scaffold protein axin together with the Ragulator complex at the lysosomal surface has been proposed as important regulators of AMPK activation (Zhang et al. 2013, 2014). These data support a model where axin bound to LKB1 recruits AMPK in the AMP-bound state, leading to AMPK phosphorylation and activation. Further, in particular under nutrient-poor conditions, the axin-LKB1-AMPK complex seems to interact with the Ragulator complex which is tethered via its LAMTOR1 component to the lysosomal surface. The Ragulator complex, apparently by its interaction with the lysosomal v-type ATPase, seems to be an independent sensor of cellular nutrient conditions. It is known to recruit the nutrient-signaling TORC1 complex (see Chap. 5) to lysosomes under nutrient-rich conditions (Bar-Peled and Sabatini 2014), thus suggesting reciprocal recruitment and activation of axin-LKB1-AMPK or mTORC1, depending on the cellular nutrient state (Hardie 2014c). It is currently unknown whether such regulation exists in the heart.
There is also some evidence that AMPK subunit isoforms determine specific protein/protein interactions. The β-subunit may in some cases confer substrate specificity, as has been shown in yeast (Vincent and Carlson 1999) and plants orthologs (Polge et al. 2008), but with mammalian AMPK [IntAct database (Kerrien et al. 2012)]. We recently found the β2-isoform interacting with Mu- and Pi-type glutathione transferases (GSTs) to favor glutathionylation of the α-subunit (Klaus et al. 2013). However, in the case of fumarate hydratase (FH), we identified a specific interaction with α2-containing AMPK complexes to facilitate FH phosphorylation (Klaus et al. 2012).
10.4 Activation
AMPK integrates various intra- and extracellular signals and maintains cross talk with other signaling pathways. This makes the kinase a central signaling hub in sensing and regulating cellular energetics and ATP-dependent functions. Indeed, the most recent research revealed that AMPK activation is much more complex than initially anticipated and that it depends on multiple covalent modifications and allosteric effectors (Fig. 10.2). Such AMPK regulation evolved from a more simple state as, e.g., in the yeast AMPK homologues that lack allosteric activation by AMP to the more complex regulation present in vertebrates.
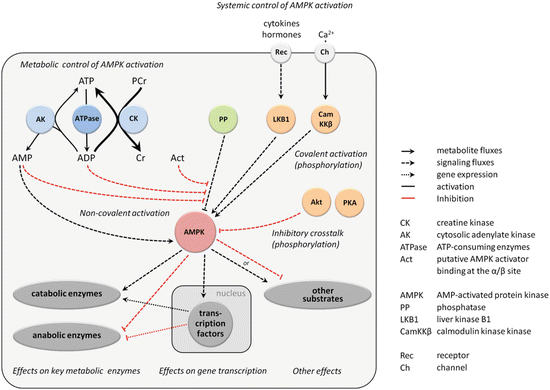
Fig. 10.2
AMPK signaling. AMPK is activated by intra- and extracellular metabolic and endocrine signals and affects various downstream processes. Activation of AMPK is triggered by upstream kinases (covalent activation by LKB1, CamKKβ, inhibition by Akt and PKA) and phosphatases. They mediate mainly extracellular signals carrying, e.g., information on the energy and nutrient state of the cellular environment and the entire organism (endocrine signals; systemic control of AMPK). Covalent activation also depends on some intracellular parameters (Ca2+, possibly also ROS/RNS), as well as the allosteric ligands. The second layer of regulation is represented by AMPK activation via AMP and ADP (allosteric regulation), both acting as second messengers of cellular energy stress (metabolic control of AMPK). This signaling is linked to conversion of nucleotides via the adenylate kinase (AK) and creatine kinase (CK) reactions. Activated AMPK compensates for ATP loss by accelerating catabolism and inhibiting anabolism and exerts further effects on cell motility, growth, proliferation, and others, via regulation of key enzymes and transcription factors. For further details see text
Covalent Regulation by Phosphorylation
The phosphorylation state of the conserved threonine within the kinase domain activation loop (conventionally referred as Thr172) determines the primary activation of AMPK. As compared to an inactive state, this phosphorylation can increase kinase activity by more than 100-fold (Suter et al. 2006). The AMPK phosphorylation state depends on the balance between the activity of different upstream kinases and phosphatases. The two well-established upstream kinases are tumor suppressor kinase complex LKB1-STRAD-MO25 (Woods et al. 2003; Shaw et al. 2004; Hawley et al. 2003) and Ca2+-calmodulin-dependent protein kinase kinases (CamKK), in particular CamKKβ (Hawley et al. 2005; Woods et al. 2005; Hurley et al. 2005). LKB1 is the major AMPK kinase in most cells, including cardiomyocytes. However, this kinase seems to mostly exhibit constitutive activity and may thus not be the limiting step in AMPK activation. More recent studies suggest that close co-localization of LKB1 with AMPK involving the scaffold protein axin and the lysosomal surface may be necessary for efficient AMPK activation via the LKB pathway (see Chap. 3; Zhang et al. 2013, 2014). In some cell types, in particular in the brain but much less in the heart, AMPK is activated predominantly in a Ca2+-dependent manner by CamKKβ. Such CamKKβ-mediated AMPK activation might anticipate an increasing energy turnover that accompanies a rise in cytosolic Ca2+ during muscle contraction, but its role in the heart is not well understood. Transforming growth factor-β (TGF-β)-activated kinase 1 (TAK1) has been suggested as another AMPK kinase (Herrero‐Martín et al. 2009) and also as an AMPK substrate (Kim et al. 2012b). TAK-1 is present in the heart, but not activated during ischemia, and it is unclear whether it acts via direct AMPK phosphorylation (Xie et al. 2006b).
Protein phosphatases are possibly the more critical parameter governing the α-Thr172 phosphorylation state, and this may also apply to the heart. AMPK covalent activation is modulated by the protein phosphatase 1 (PP1) (Garcia-Haro et al. 2010), the protein phosphatase 2C (PP2Cα) (Sanders et al. 2007a), and the calcium-mediated protein phosphatase 2A (PP2A) (Park et al. 2013). It was also proposed that α-SNAP may exhibit phosphatase activity on AMPK Thr172 according to in vitro dephosphorylation assay (Wang and Brautigan 2013). However, in tissues including the heart and endothelial cells, especially expression levels PP2C and 2A, respectively, correlate well with AMPK activation (Wang and Unger 2005; Wu et al. 2007).
There seems to be a cross talk of AMPK with many other cellular signaling pathways. Mainly the α-Thr172 phosphorylation state is negatively controlled by hierarchical phosphorylation at other sites in the AMPK heterotrimer, in particular in the α-Ser/Thr-rich loop. Protein kinase B (PKB/Akt) that is activated under glucose-rich conditions by insulin signaling is inhibiting AMPK by phosphorylation at rat α1-Ser485 (much less so at the equivalent α2-Ser491) which reduces phosphorylation at the activating α-Thr172 (Hawley et al. 2014; Horman et al. 2006). Thus, hyperactivation of PKB/Akt as occurring in many tumor cells, and also in the heart under doxorubicin treatment (see Chap. 6), can strongly downregulate AMPK activation, with negative effects on proliferation control and cell energetics, respectively. Similar inhibitory phosphorylations of AMPK were reported for protein kinase GSK3β (Suzuki et al. 2013) and protein kinase A (PKA) (Hurley et al. 2006; Djouder et al. 2010). In the latter case, not the Ser/Thr-loop phosphorylations (including Ser485/Ser491) seem to be inhibitory but rather another one at α-Ser173 (Djouder et al. 2010). The physiological rationale underlying AMPK inhibition by GSK3β and PKA is, however, less obvious. AMPK is further negatively controlled by the Ras/Raf/MEK/ERK pathway in a more complex manner, involving negative feedback loops. While active AMPK can reduce MEK/ERK signaling via phosphorylation of upstream B-Raf (Shen et al. 2013), active ERK can reduce AMPK signaling by inhibitory phosphorylation of the AMPK upstream kinase LKB1 (Zheng et al. 2009). Further phosphorylation sites were identified in both AMPK α- and β-subunits, many of them targeted by autophosphorylation, but their functional role remains uncertain. As recently discovered, (auto)phosphorylation of β-Ser108 close to the glycogen-binding domain seems to be an important second allosteric regulatory site (see below).
Endocrine Signals
Information about the cellular environment and whole-body energy and nutrient state is linked to AMPK signaling via endocrine, paracrine, and autocrine mechanisms. These include a diverse array of hormones and cytokines (Table 10.2). They regulate AMPK mainly by triggering AMPK phosphorylation via upstream kinases, and this regulation often occurs in a tissue-specific manner. Best studied are probably the orexigenic/anorexigenic hormones ghrelin and leptin. In peripheral tissues, leptin activates AMPK to regulate fatty acid oxidation and glucose uptake. In hypothalamus, leptin inhibits and ghrelin activates AMPK to decrease and increase appetite, respectively, in order to regulate food intake (Steinberg and Kemp 2009; Steinberg 2013). Other endocrine factors that affect AMPK activity are sex hormones that act via LKB1 (McInnes et al. 2012) and angiotensin 2 (Nagata et al. 2004; Steinberg 2013). Endocrine signals in the heart are discussed in Chap. 6.
Table 10.2
Hormones and cytokines affecting AMPK activity
Compound | Effect | Mechanism | Tissue | Ref. |
|---|---|---|---|---|
Leptin | + | AMP increase | Muscle | Minokoshi et al. (2002) |
Leptin | − | Melanocortin receptor signaling? | Hypothalamus | Minokoshi et al. (2004) |
Interleukin-6 | + | Not known | Muscle | Carey et al. (2006) |
Tumor necrosis factor α | − | Increased PP2C expression | Muscle | Steinberg et al. (2006) |
Resistin | − | Not known | Liver, muscle, adipose | Banerjee et al. (2004) |
Ghrelin | + | G protein coupled receptor signaling CamKK activation | Hypothalamus, heart | |
Ghrelin | − | Liver | Barazzoni et al. (2005) | |
Adiponectin | + | Adiponectin receptor 1 signaling? | Muscle, adipose, hypothalamus | Kubota et al. (2007) |
Estrogen | + | Not known | Muscle | D’Eon et al. (2005) |
Testosterone; dihydrotestosterone | − | Decrease in LKB1 mRNA | Adipocytes | McInnes et al. (2012) |
17β-estradiol | + | Increase in LKB1 mRNA | Adipocytes | McInnes et al. (2012) |
Angiotensin 2 | + | AT1R-NADPH oxidase axis | Vascular smooth muscle cells | Nagata et al. (2004) |
Calcium Signals
As described above, cellular calcium can regulate the Thr172 phosphorylation state of AMPK via calcium-homeostasis-related kinases in phosphatases, in particular CamKKβ (Hawley et al. 2005) and PP2A (Park et al. 2013), respectively. This can also be demonstrated with calcium ionophores (e.g., A23187) in LKB1-deficient cells. Ca2+– and AMP-dependent AMPK activation occurs independently and can be synergistic, since AMP binding (see below) protects the Ca2+-induced phosphorylation (Fogarty et al. 2010).
Non-covalent Regulation
The second major mechanism of AMPK activation relies on non-covalent, allosteric regulation. It mainly occurs by AMP and ADP, competing with MgATP for binding to the γ-subunit CBS domains. At a low cellular energy state, increases of AMP and, as discovered more recently, also of ADP can be sensed by AMPK as altered AMP/ATP and ADP/ATP concentration ratios (Oakhill et al. 2011; Xiao et al. 2011). In many cell types and in particular in the heart and skeletal muscle, breakdown of ATP to ADP at the onset of high workload or cellular stress has only minor immediate effects on ATP levels. Due to the energy buffer and transfer function of the CK/PCr system, global and local ATP pools are rapidly replenished (Schlattner et al. 2006; Wallimann et al. 2011). Thus, ATP is not a very suitable signal for indicating developing energy deficits. However, minor decreases in ATP levels lead to more pronounced relative increases in free ADP and even more in AMP due to the adenylate kinase (AK) reaction. Under these conditions, AK uses two ADPs to regenerate ATP and AMP, thus increasing AMP concentrations from the sub-micromolar range under resting conditions to the lower micromolar range (Hardie et al. 2011). To a lesser extent, AMP levels also depend on pyrophosphates (cleaving the β-phosphate bond of ATP) and the activity of AMP degradation pathways [AMP-deaminase and 5′-nucleotidase, whose inhibition may be useful to activate AMPK (Kulkarni et al. 2011)]. As a consequence, a decrease in ATP levels by only 10 % translates into a ten- to 100-fold increase in AMP, making AMP an ideal second messenger of energy stress. Regulation of AMPK activation by the balance between ATP, ADP, and AMP concentrations resembles to what was put forward by Atkinson 50 years ago as “energy charge” regulation (Atkinson 1968; Hardie and Hawley 2001; Xiao et al. 2011; Oakhill et al. 2011).
The molecular basis of AMPK activation by AMP and ADP is not yet fully understood but involves binding to CBS sites on the γ-subunit that trigger multiple interconnected mechanisms. Binding of AMP leads to an up to a ~tenfold allosteric activation of AMPK (Gowans et al. 2013). Earlier in vitro studies suggested that the α2-subunit has a higher sensitivity to this allosteric activation (Salt et al. 1998a). In addition, AMP and ADP binding increase the phosphorylation status of α-Thr172 through protection of the α-subunit activation loop from dephosphorylation by phosphatases (Davies et al. 1995; Xiao et al. 2011). In addition, AMP (but not ADP) promotes α-Thr172 phosphorylation by LKB1 but not by CamKKβ (Gowans et al. 2013). The γ-subunit CBS sites involved in these allosteric effects are sites 1, 3, and 4. However, there is some debate on the role of these sites, in particular which sites mediate direct allosteric activation and which ones the protection of dephosphorylation. There is a consensus that changes of AMP and ADP concentrations in the physiological range are mainly sensed at sites 1 and 3, called exchangeable binding sites. Here, free AMP and ADP probably compete mainly with free ATP, since the most abundant Mg2+-complexed ATP has tenfold lower affinity for the CBS sites (Xiao et al. 2011). Sites 1 and 3 differ about 30-fold in their affinity for adenylates, and initial evidence suggested site 1 as high-affinity site, sensing low micromolar concentrations of AMP for allosteric activation, and site 3 as low affinity site, involved in protection of dephosphorylation at higher AMP and ADP concentrations (Xiao et al. 2011). However, the role of CBS sites may not be defined as clearly. A more recent study suggests that site 3 is the most important for allosteric activation (Chen et al. 2012). Indeed, mutation of site 3 residues abrogates allosteric AMPK activation (Chen et al. 2012; Scott et al. 2004), and this site is also in contact with the α-subunit (see below). In addition, site 4 may play a role in allosteric activation. This is a tight AMP-binding site, generally reported as non-exchangeable site since purified protein or protein crystals always retain AMP in this site, even when treated with ATP. However, Chen et al. (Chen et al. 2012) observed ATP at site 4 when co-crystallizing AMPK core complex in the presence of 2 mM free ATP, a very high concentration that may not be physiologically relevant. However, ATP binding to site 4 forces site 3 to remain empty, and this affects allosteric AMPK activation, consistent with the model of CBS site 3 being the major site of allosteric regulation. A complicating fact is that some nucleotide-binding CBS residues can interact with nucleotides at different sites, thus precluding a clear-cut functional assignment of CBS sites (Hardie 2014c).
All known direct AMPK activators act via allosteric effects (see Chap. 7). They either act like AMP at the CBS sites (e.g., 5-aminoimidazole-4-carboxamide riboside, AICAR; Giri et al. 2004) or they exert their effects by binding to an entirely different site, discovered only recently (e.g., A-769662; Scott et al. 2008). This site is situated in a cleft between the α-kinase domain and the β-CBM domain and stabilized by autophosphorylation of the β-Ser108. Occupation of this α/β site confers protection of dephosphorylation. It can be speculated that there exists an endogenous activating metabolite binding at the α/β site, and/or an endogenous activating kinase, able to phosphorylate Ser108 (Hardie 2014c).
All these allosteric mechanisms, whether they involve binding events at the CBS sites or at the novel α/β site, require close communication between the sensing subunit (γ or β) and the catalytic subunit (α). We and our collaborators have proposed that subunit communication and activation occur via a conformational switch within the AMPK full-length complex (Riek et al. 2008; Chen et al. 2012). Indeed, AMP-induced conformational changes have been evidenced through structural studies by SAXS (Riek et al. 2008), electron microscopy (Zhu et al. 2011), and X-ray crystallography (Chen et al. 2012) within different parts of the AMPK heterotrimer. Recent structures of the holo-AMPK complex in its active state, as well as low-resolution structures in Thr172 phosphorylated and unphosphorylated states, suggest that conformational changes and intramolecular movements involve α-RIM, α-AID, the two lobes of the α-kinase domain, as well as the entire γ-subunit (Chen et al. 2013; Xiao et al. 2011; Calabrese et al. 2014; Li et al. 2014). High-resolution apo-AMPK structures of holo-AMPK complex will be necessary to answer the remaining questions, in particular how a different occupation of CBS sites communicates via α-RIM and α-AID with the kinase domain. Collectively, these non-covalent AMPK activation mechanisms add an important layer to the regulation of AMPK activity, since they allow a direct response to intracellular metabolites.
Exercise and Hypoxia
Given the sensitivity of AMPK for adenine nucleotides, any physiological or pathological situation that changes adenylate ratios will affect AMPK signaling. AMPK is activated by a plethora of stimuli such as metabolic stresses and drugs and xenobiotics that either (1) inhibit ATP production, such as starvation for glucose (Salt et al. 1998b) and oxygen (Marsin et al. 2002), or metabolic poisons or (2) increase ATP consumption, such as muscle contraction (Lantier et al. 2014). Muscle contraction and exercise in general trigger rapid activation of AMPK (Chen et al. 2003), and this may be one of the fastest mechanisms that mediate metabolic adaptation to exercise. When AMPK is knocked out in the skeletal muscle of β1β2 transgenic mice, they lose exercise tolerance and glucose uptake during contractions, become physically inactive, and present an importantly impaired capacity for running linked to reductions in skeletal muscle mitochondrial content (O’Neill et al. 2011). During hypoxia, from the early stage on, a drastic drop in the ATP/AMP level occurs, resulting in AMPK activation.
Other Covalent and Non-covalent Regulations
In addition to the above-described conventional regulation of AMPK, there is increasing evidence for additional activation and inactivation mechanisms. Here, different secondary protein modifications play an important role. Myristoylation at Gly2 in the β-subunit increases the sensitivity of AMPK for allosteric activation and promotes Thr172 phosphorylation (Oakhill et al. 2010). The β2-subunit, but not β1, is sumoylated by the E3-small ubiquitin-like modifier (SUMO) ligase protein inhibitor of activated STAT (PIASy), which attaches SUMO2 but not SUMO1 moieties. This seems to enhance AMPK activity and competes with ubiquitination that results in inactivation of AMPK complex (Rubio et al. 2013). Ubiquitination of AMPK occurs via complexes of laforin (a dual-specificity protein phosphatase) and malin (an E3-ubiquitin ligase), mainly at the β-subunit, and leads to K63-linked ubiquitin chains that are involved in functions different from proteasome degradation (Moreno et al. 2010). Glutathionylation at Cys299 and Cys304 in the α-subunit activates the kinase under oxidative conditions in cellular models and is favored by binding to certain GST isoforms (Klaus et al. 2013). This latter mechanism may be part of a more general redox regulation of the kinase (Han et al. 2010; Jeon et al. 2012). ROS and RNS activate AMPK, but it is unclear whether this happens via increases in ADP and AMP concentrations or whether noncanonical mechanisms at the level of AMPK (like glutathionylation) or upstream kinases play a role. Vice versa, AMPK regulates NADPH homeostasis and an entire battery of ROS-detoxifying enzymes. Another non-covalent allosteric regulator is glycogen as well as other synthetic branched oligosaccharides that inhibit AMPK activity by binding to the β-CBM domain (McBride et al. 2009) (see above).
10.5 Regulation
Metabolism
AMPK regulates cellular metabolism at many levels, reducing anabolism (ATP-demanding processes) and upregulating catabolism (ATP-generating processes) to restore a healthy energy status at a cellular and whole-body level. To do so, AMPK directly acts on metabolic key enzymes and signaling proteins (acute effects) or on transcription factors (chronic effects, see Fig. 10.2) (Hardie et al. 2012b). Interestingly, drugs of the two main classes of antidiabetic drugs, biguanides (e.g., metformin) and thiazolidinediones (e.g., rosiglitazone), both act at least in part through activation of AMPK (Morrison et al. 2011; Musi et al. 2002). In the heart, AMPK is part of the signaling network that allows a predominant use of fatty acid oxidation for ATP generation and also provides the metabolic flexibility to respond to changes in substrate availability, thus continuously matching ATP generation and demand. Failing of AMPK to provide this flexibility under certain pathological conditions can contribute to the pathogenesis of heart failure (see Chap. 6, reviewed in Kim and Dyck 2015).
Lipid Metabolism
Activated AMPK induces transfer of fatty acid transporter (FAT/CD36) to the plasma membrane to increase fatty acid uptake (Luiken et al. 2003). AMPK further inhibits ATP-consuming lipid synthesis, notably in the liver and in the adipose tissue, but stimulates lipid catabolism for ATP generation. Phosphorylation of acetyl-CoA carboxylase (ACC) decreases ACC-catalyzed formation of malonylCoA a precursor in the fatty acid synthesis pathway. At the same time, reducing malonylCoA levels will relieve their inhibition of carnitine palmitoyltransferase 1 (CPT-1), which triggers fatty acid import into mitochondria and subsequent β-oxidation. AMPK also phosphorylates and inhibits other anabolic enzymes: 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR), a key enzyme in cholesterol synthesis that converts 3-hydroxy-3-methylglutaryl-CoA into mevalonic acid, and glycerol phosphate acyltransferase, involved in triglyceride and phospholipid synthesis (Liao et al. 2014). Since AMPK acts by stimulating lipolysis and inhibiting lipogenesis, its pharmacological activation seems to be useful to treat obesity, diabetes type 2, and more generally the metabolic syndrome (Hardie 2008a; O’Neill et al. 2013).
Carbohydrate Metabolism
AMPK also interferes with carbohydrate metabolism at different levels, including carbohydrate uptake, glycolysis, and glycogen synthesis. Activated AMPK promotes cellular glucose uptake via glucose transporters GLUT1 (expressed in most cells except muscle, liver, and adipose tissue) and GLUT4 (expressed mainly in adipose tissue and striated muscle). AMPK activation promotes GLUT4 translocation to the plasma membrane (Kurth-Kraczek et al. 1999) and stimulates GLUT4 transcription by phosphorylation of the transcription repressor histone deacetylase 5 (HDAC5) which reduces its affinity for the GLUT4 promoter (McGee et al. 2008). GLUT1-dependent glucose uptake is activated via an unclear mechanism that involves GLUT1 already located at the plasma membrane (Barnes et al. 2002). Notably in case of energy deprivation in the heart, AMPK phosphorylates and activates the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB) to increase the steady-state concentration of fructose-2,6-bisphosphate (Marsin et al. 2000). This metabolite then acts as an allosteric activator of glycolysis by stimulating the glycolytic enzyme 6-phosphofructo-1-kinase (PFK1), a rate-limiting glycolytic enzyme. Once activated, AMPK also represses anabolic glucose storage into glycogen by directly phosphorylating and inactivating glycogen synthase (Bultot et al. 2012). Finally, AMPK affects carbohydrate metabolism indirectly by phosphorylation of the mTor–raptor complex, which was proposed to modulate insulin sensitivity by regulating protein levels of IRS-1 (Haruta et al. 2000; Kahn et al. 2005).
Transcription
AMPK phosphorylates and regulates various transcription factors and coregulators, including forkhead box O (FoxO) proteins FoxO1 and FoxO3 (Kubli and Gustafsson 2014) and peroxisome proliferator-activated receptor (PPAR) γ coactivator-1a (PGC-1α) (Patten and Arany 2012), both having important roles in the regulation of cardiac energetic homeostasis and beyond. PGC-1α is a central transcriptional coactivator that orchestrates mitochondrial biogenesis and dynamics, fuel transport and/or consumption, angiogenesis, and antioxidative effects. PGC-1α phosphorylation by AMPK results in improved metabolism of fatty acids and more efficient energy utilization (Schilling and Kelly 2011). FoxO transcription factors regulate expression of genes involved in the antioxidative stress response and in the balance between apoptosis, autophagy, and energy metabolism. These functions are critical for cardiac function (Ronnebaum and Patterson 2010). FoxO-regulated genes also encode proteins that contribute to improved energy metabolism, including FAT/CD36 and GLUT4 indirect metabolic effects.
Growth and Proliferation
Many effects of AMPK on cell growth, cell cycle, and autophagy are mediated by another evolutionary conserved serine/threonine protein kinase further downstream, the mammalian target of rapamycin (mTor). mTor occurs as two functional multiprotein complexes, mTORC1 and mTORC2 (Loewith et al. 2002). mTORC1 comprises mTOR, raptor, mLST8, and PRAS40 and is regulated by cellular energy and nutrient state, whereas mTORC2 is not. Raptor also plays a significant role in intracellular localization of mTORC1 in response to amino acid availability, which is an essential cellular signal for mTORC1 activation (Sancak et al. 2008). Activation of mTORC1 occurs at the lysosomal surface as a part of complex, multiprotein assemblies (Bar-Peled and Sabatini 2014). Active mTORC1 stimulates several ATP-demanding cellular processes such as translation, transcription (protein synthesis), ribosome biogenesis, mitochondrial metabolism, proliferation, and autophagy. Unlike mTORC2, mTORC1 is sensitive to rapamycin, a molecule used as immunorepressor due to its capacity to downregulate protein synthesis, notably of antibodies.
Two important substrates of mTORC1 in its response to nutrients and cellular energy status are S6 kinase (S6K) and eIF4E binding proteins (4EBPs). Raptor, a component of mTORC1, functions as a scaffolding protein to recruit such substrates for phosphorylation (Nojima et al. 2003). S6K is a ribosomal kinase regulating translation initiation, mRNA processing, and cell growth and notably enhances protein synthesis once phosphorylated. 4EBPs are translational repressors that are inactivated upon phosphorylation by mTORC1. To precisely regulate these mTORC1-dependent, energy-demanding processes, AMPK inhibits mTORC1 signaling through two distinct mechanisms (Inoki et al. 2012). First, it directly phosphorylates raptor at the conserved Ser722 and Ser792, leading to recruitment of 14-3-3 protein and an inactive mTORC1 complex (Gwinn et al. 2008). Second, it phosphorylates tuberous sclerosis protein 2 (TSC2), a GTPase-activating protein (GAP), thus stimulating the downstream GTPase Ras homologue enriched in brain protein (Rheb). This transforms Rheb from its GTP-bound form that activates mTORC1 into its inactive, GDP-bound form (Inoki et al. 2003). This latter pathway of mTORC1 regulation by AMPK may depend on cell type and tissue (Wolff et al. 2011). Collectively, the AMPK and mTORC1 pathways serve as a signaling nexus to regulate cellular metabolism, energy homeostasis, and cell growth. Disorder of each pathway may strongly contribute to the development of pathologies such as type II diabetes or cancer.
Autophagy and Apoptosis
While AMPK activation by upstream kinases is well studied, much less is known about regulation of AMPK stability and activity by components of the ubiquitin–proteasome system, responsible for cellular recognition and degradation of proteins. Growing evidence suggests that AMPK regulates overall proteasome activity and individual components of the ubiquitin–proteasome system (Ronnebaum et al. 2014). Autophagy is important for maintaining homeostasis when nutrient supply becomes limiting. It is important for the cellular turnover of proteins and organelles and is rapidly upregulated during stress. In metabolic disorders including obesity and diabetes, autophagy is reduced, leading to accumulation of protein aggregates and dysfunctional organelles which can contribute to pathogenesis.
10.6 Cardiac Signaling in Health and Disease
Cardiac AMPK activity is increased by many stimuli, acting either via upstream kinases or modulation of adenylate levels under both pathological and physiological stress and involving various hormones and cytokines (Fig. 10.2; Zaha and Young 2012). However, within the physiological range, the role of cardiomyocyte AMPK is possibly different from other cell types, mainly because of the remarkable metabolic stability of this organ maintained by multiple other mechanisms. Two classical physiological stimuli of AMPK, exercise and hypoxia, also act on cardiac AMPK (Coven et al. 2003; Frederich et al. 2005; Musi et al. 2005) and promote the metabolism of glucose and fatty acids via its different downstream targets. However, it is unclear whether this activation is due to altered energy state as in the skeletal muscle or rather relies on alternative upstream signaling. At least one other physiological AMPK stimulus, nutrient deprivation, seems not to operate in a canonical manner in the heart (Clark et al. 2004). Apart of these key roles, cardiac AMPK mediates the cardiomyocyte response to a variety of other physiological or pathological situations, including some forms of pressure overload, heart failure, intracellular calcium overload, or reactive oxygen and nitrogen species (Dolinsky et al. 2009; Zaha and Young 2012).
Ischemia
As a pathological stimulus, ischemia is the best studied both in form of no-flow and partial ischemia in isolated perfused animal hearts, as well as regional ischemia due to coronary ligation in vivo (Kim et al. 2011; Kudo et al. 1996; Paiva et al. 2011; Russell et al. 2004; Wang et al. 2009), for a review see Young (2008). They lead to rapid and lasting AMPK activation. As already mentioned, oxidative stress may be a determinant of such activation, acting through different forms of ROS (Sartoretto et al. 2011; Zou et al. 2002). In endothelial cells, it is rather peroxynitrite formation that affects AMPK via the protein kinase Cζ-LKB1 axis (Xie et al. 2006a; Zou et al. 2004), while in other non-excitable cells, it may be rather an ROS-induced Ca2+ release that triggers the CamKKβ axis (Mungai et al. 2011). ROS-facilitated glutathionylation of AMPK (see Chap. 4) as observed in cellular systems represents yet another direct activation mechanism but still has to be verified in cardiomyocytes (Klaus et al. 2013; Zmijewski et al. 2010). However, the signaling function of ROS may be lost at more intense oxidative stress that inactivates AMPK (Gratia et al. 2012). Stress resulting from many but not all forms of pressure overload also results in AMPK activation, mainly increasing glucose uptake and glycolysis (Allard et al. 2007; Li et al. 2007; Tian et al. 2001; Zhang et al. 2008), as well as changing the gene expression profile (Hu et al. 2011).
Endocrine Regulation
Cardiac AMPK is also regulated by extracellular signals as adiponectin (Shibata et al. 2004), leptin (Minokoshi et al. 2002), resistin (Kang et al. 2011), ghrelin (Kola et al. 2005), IL6 (Kelly et al. 2004), and CNTF (Watt et al. 2006). Best studied are probably the hormones adiponectin and leptin (the latter at least during ischemia; McGaffin et al. 2009) which activate cardiac AMPK or the proinflammatory cytokine IL-6 which appears to reduce AMPK content and activity (Zaha and Young 2012). Cardiac AMPK seems to be involved in the positive effects of adiponectin for cardioprotection during ischemia and for reduced cardiac hypertrophy (Shibata et al. 2004, 2005). Also, leptin may modulate AMPK in the heart, since impaired leptin signaling correlates with reduced AMPK activation and metabolic defects or reduced postconditioning after ischemia (Bouhidel et al. 2008; McGaffin et al. 2009). Proinflammatory cytokines like IL-6 rather reduce AMPK protein and activation (Ko et al. 2009), although there may be opposite effects in specific tissues like skeletal muscle due to a specific autocrine–paracrine effect (Kelly et al. 2004). Another cytokine with functions in the heart is macrophage migration inhibitory factor (MIF), which is involved in AMPK activation during ischemia and hypoxia, and its decrease with age in mice seems to reduce AMPK activation during ischemia (Ma et al. 2010; Miller et al. 2008).
Protein Turnover
AMPK has been suggested to play an important role in regulating cardiac turnover of proteins and organelles (Baskin and Taegtmeyer 2011; Zaha and Young 2012), also during ischemia. This process is critical for the survival and self-renewal of terminally differentiated cells as cardiomyocytes and requires tightly regulated degradation of misfolded and damaged proteins or damaged/dysfunctional organelles (as, e.g., mitochondria) and their replacement by new and functional entities. Recent evidence suggests that AMPK regulates degradation at two levels. Individual proteins are eliminated by the ubiquitin–proteasome system, with AMPK activating the cardiac ubiquitin ligases atrogin-1 and MuRF. Whole organelles are digested by stimulation of autophagy via activation of ULK1 and inhibition of mTOR (Baskin and Taegtmeyer 2011; Hardie et al. 2012a; Zaha and Young 2012). Thus, under conditions of metabolic stress, AMPK activation inhibits protein synthesis (via mTOR) and activates degradation of proteins and organelles. The recycling of nutrients from breakdown of cellular components (macromolecules and organelles) contributes to the maintenance of the cellular ATP-regenerating capacity, to the control of protein and organellar quality, as well as to the maintenance of cardiomyocyte size and their survival. It is to note that slowing down protein synthesis also prevents accumulation of unfolded proteins under stress situation such as hypoxic or ischemic injury and the related endoplasmic reticulum stress (Terai et al. 2005).
Inflammation
As mentioned above, cytokines can directly regulate cardiac AMPK activity. On the other hand, AMPK has the capacity to repress inflammatory responses and exert anti-inflammatory and immunosuppressive effects in a variety of cell types by interfering with cytokine signaling (Salminen et al. 2011; Salt and Palmer 2012). There is evidence that in several tissues, including the cardiovascular system, activation of AMPK impairs leukocyte infiltration (an early key step in development of inflammation) by reducing expression of chemokines and adhesion molecules (Salt and Palmer 2012). This is important for the heart, since inflammation is a critical component in the pathogenesis of many common cardiovascular diseases (Pankuweit et al. 2004), including the diabetic heart (Ko et al. 2009).
Cardioprotection
Most mechanisms triggered by active cardiac AMPK, though possibly not all, are recognized to promote cardioprotective effects. For example, AMPK-dependent stimulation of glucose metabolism (glucose uptake by GLUT4 and stimulated glycolysis by PFK2) is of particular importance for the anaerobic ATP synthesis during ischemia and thus for protection of the ischemic heart (Young 2008). It is to note, however, that AMPK activation persisting after ischemia during early reperfusion is considered rather detrimental, because excessive stimulation of fatty acid oxidation impairs glucose oxidation via Randle cycle/uncoupling of enhanced glycolysis from glucose oxidation (Dyck and Lopaschuk 2006). The net result of AMPK activation during an ischemia/reperfusion episode can still be considered as beneficial (Zaha and Young 2012). Finally, AMPK was suggested to mediate the cardiac response to different known cardiac protectants, mostly in pathological setting, as, e.g., during ischemic episodes or pathological hypertrophy (review in Kim et al. 2009). For example, AMPK contributes to the cardioprotective effects of adiponectin and metformin during coronary occlusion in mice (Calvert et al. 2008; Shibata et al. 2005), as well as to cardiac preconditioning by regulating the activity and recruitment of sarcolemmal K(ATP) channels (Sukhodub et al. 2007).
As AMPK activation has predominantly pro-survival character, it is considered as promising potential therapeutic target in the treatment of different cardiovascular diseases (Inoki et al. 2012; Kim et al. 2011; Zaha and Young 2012).
Cardiomyopathies
Mutation of specific CBS residues is associated with pathological disorders (Kemp 2004; Ignoul and Eggermont 2005). Mutations in the CBS domains of the AMPK γ2-subunit, expressed at particularly high levels in the heart, cause the Wolff-Parkinson-White (WPW) syndrome, a hereditary cardiomyopathy of varying severity, involving cardiac hypertrophy, contractile dysfunction, and arrhythmias. Mutations impair adenylate binding and thus AMPK activation (Scott et al. 2004; Burwinkel et al. 2005), but the major cause for the cardiomyopathy is the increased AMP-independent basal AMPK activity. This leads to higher glucose uptake, accumulation of glycogen in cardiac myocytes, and finally impairment of heart muscle development (Burwinkel et al. 2005; Davies et al. 2006).
Cardiac Contractility
Cardiac troponin I was identified in a yeast 2-hybrid screen to interact with the AMPK γ2-subunit N-terminal domain and to be phosphorylated by AMPK at Ser150 in vitro and during ischemia in the heart (Oliveira et al. 2012). This results in increased myocyte contraction and prolonged relaxation by an increase in myofilament Ca2+ sensitivity. These effects were also triggered by the AMPK activator AICAR, suggesting that pharmacological AMPK activation could improve heart function.
Doxorubicin-Induced Cardiotoxicity
The anthracycline antibiotic doxorubicin (Adriamycin; DXR) remains one of the most largely prescribed chemotherapeutic drugs for the treatment of a variety of human cancers (Eschenhagen et al. 2011; Ewer and Ewer 2010; Gianni et al. 2008; Minotti et al. 2004, 2010). It is still a cornerstone of combination therapies together with more targeted, new generation drugs. Unfortunately, the potent antitumor effect of DXR is accompanied by a number of unwanted side effects, in particular a serious cardiac toxicity. This complication represents a major obstacle when using the drug for prolonged time and/or at a higher cumulative dose (Curigliano et al. 2012; Menna et al. 2008). Detrimental effects of DXR are thought to be mediated by different kinds of stress induced by the drug: energetic stress, oxidative stress, and genotoxic stress (Gratia et al. 2012). Given the stress-sensing function of AMPK, an activation of the kinase is expected in the heart as a result of drug action. Paradoxically, it seems that in the heart DXR does not increase but rather decrease the basal phosphorylation of AMPK, thus inactivating the kinase. Such AMPK inhibition has been observed in different model systems of acute and chronic cardiotoxicity, including cultured cardiomyocytes (Konishi et al. 2011; Wang et al. 2012a), perfused heart (Gratia et al. 2012; Tokarska-Schlattner et al. 2005), and in vivo models of rat (Cai et al. 2010; Gratia et al. 2012; Russell 2003) and mice (Kawaguchi et al. 2012; Kim et al. 2010; Konishi et al. 2011). AMPK appears to be an early and sensitive DXR target: in rat hearts perfused with rather low clinically relevant DXR concentrations, AMPK is inhibited already after 1–2 h, well before changes in myocardial function can be observed. In rats, AMPK inactivation persists several weeks after the end of treatment (Gratia et al. 2012; Konishi et al. 2011). Thus, it seems that DXR generates conditions which normally should activate AMPK but instead inhibits the stress response of AMPK. This may create a vicious cycle for the heart, important for drug toxicity.
DXR-induced inhibition of cardiac AMPK signaling is, at least partially, due to the negative cross talk with other signaling pathways, in particular Akt and ERK. These two pro-survival kinases respond in the heart to a variety of stimuli (Kehat and Molkentin 2010; Sussman et al. 2011) and are activated by DXR (Gabrielson et al. 2007; Gratia et al. 2012; Horie et al. 2010; Khalil et al. 2012; Kobayashi et al. 2010; Lee et al. 2006; Lou et al. 2005; Tokarska-Schlattner et al. 2010). Both kinases are known to inhibit AMPK (Du et al. 2008; Esteve-Puig et al. 2009; Hahn-Windgassen et al. 2005; Horman et al. 2006; Kovacic et al. 2003; Soltys et al. 2006).
This interplay of AMPK with Akt seems especially pronounced in heart, and AMPK inhibition by the Akt pathway has been also reported for other cardiac pathologies (Dyck and Lopaschuk 2006). We could substantiate the role of the Akt–AMPK cross talk for drug-induced AMPK inhibition by using the specific Akt inhibitor MK2206 (Gratia et al. 2012). Akt is mainly activated by DNA-damage signaling in response to strong DNA damage that is induced in cardiomyocytes by the drug. This involves DNA-dependent protein kinase (DNA-PK), activated by DNA double-strand breaks which are a typical consequence of DXR action. Interestingly, several most recent studies indicate a more general relationship between a reduced LKB1–AMPK signaling and cardiac disease (Dolinsky et al. 2009; Ikeda et al. 2009; Shaw 2009). Decreased activation state of the LKB1–AMPK axis occurs in several cardiac pathologies, and in some of them a similar AMPK inhibition by cross talk with Akt has been suggested as underlying mechanism (Hahn-Windgassen et al. 2005; Horman et al. 2006; Kovacic et al. 2003; Soltys et al. 2006). In spontaneously hypertensive rats, another mechanism has been put forward, namely, oxidative damage of LKB1 due to formation of adducts between 4-hydroxy-2-nonenal (HNE, product of lipid peroxidation) and LKB1 which inhibits LKB1 and thus also AMPK activity (Dolinsky et al. 2009). Taken together, these data suggest that AMPK activation as a potential preventive or therapeutic strategy during DXR treatments.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


