Fig. 8.1
Morphology and distribution of alveolar epithelial cells. The en face representation of alveolar epithelium shows the relative size and number of type I and type II cells [115]. The monolayer is heterogeneous and predominantly contains two classes of cell–cell interfaces: type I–type I cell junctions and type I–type II cell junctions. Adapted from [26]
Critically, Type I and Type II cells function in a cohesive manner in order to regulate alveolar fluid balance. This depends on functional interactions between these cells, which are regulated in large part by intercellular junctions at sites where cells are in direct contact. Paracrine signaling by cytokines and extracellular ATP also integrate alveolar epithelial cell function.
The alcoholic lung presents a unique pre-disease state that renders the tissue susceptible to damage when presented with a significant inflammatory insult. This is in large part because the ability of the lung to maintain a proper air/liquid interface is compromised due to a deficit in alveolar epithelial barrier function. Here we focus on how alcohol impairs alveolar epithelial physiology, with particular emphasis on that critical barrier function.
Effects of Alcohol on Alveolar Glutathione
Lung airspaces are directly exposed to environmental oxygen, which makes them vulnerable to oxidant stress. In patients requiring oxygen supplementation, there is even greater oxidant stress. As a major component of antioxidant defense, lungs use the tri-peptide thiol antioxidant glutathione [1]. Levels of glutathione in the alveoli are among the highest in the human body and are present at concentrations of 400 μM or more in the alveolar fluid. Type II cells also contain high levels of intracellular mitochondrial and cytosolic glutathione pools that enable them to resist oxidant stress.
In contrast to the unstressed lung, chronic alcohol ingestion increases oxidative damage by depleting glutathione [2, 3]. Type II cells from alcohol-fed rats contain 20 times less total glutathione than controls and airspace glutathione is depleted [2, 4]. Glutathione levels are further decreased during endotoxemia where whole lung glutathione was reduced by 25 % in alcohol-fed septic rats as compared to control-fed septic rats, consistent with an additive effect of sepsis as a second hit on oxidative stress when it occurs in alcoholics [5, 6].
Metabolsim of dietary alcohol (ethanol) to acetaldehyde directly depletes glutathione, causing oxidant stress [6, 7]. Alcohol also induces cell-signaling pathways that contribute to oxidant stress, including increasing the production of Transforming Growth Factor β (TGF-β), which appears to play a critical role in mediating the alcoholic lung phenotype [5, 8]. TGF-β decreases lung glutathione by inhibiting γ-glutamylcysteine synthetase expression [9, 10] and further increases oxidant stress by increasing Nox expression [11, 12] and H2O2 production [13]. Consistent with this effect on oxidant stress, TGF-β accelerates the progression of acute lung injury [14, 15].
The effects of alcohol on the total cell glutathione pool suggest the potential of antioxidant therapies as a means to reduce the severity of acute lung injury. Consistent with this, supplementing the diets of alcohol-fed rats with the glutathione precursor procysteine reduces oxidant stress and reverses many of the pathophysiological effects of alcohol on the lung [16–18]. Antioxidant therapies have not yet been proven to be effective in decreasing mortality in critically ill patients since they have never been directly tested in individuals with alcohol use disorders. However, antioxidants can prevent the severity and duration of acute lung injury [19, 20]. Therefore, bolstering the antioxidant reserves of the lung are likely to be best used as an adjunct to other therapeutic approaches.
Alveolar Epithelial Barrier Function and Fluid Balance
The alveolar epithelium controls fluid balance through a combination of processes. First, the alveolus provides a permeability barrier which prevents fluid efflux from the bloodstream, lymphatic system, and tissue into the airspaces. Second, the alveolus actively promotes fluid efflux from the airspaces through a system of ion channels where water osmotically accompanies ion transport. To some extent, barrier function and ion transport can compensate for each other; however, the extent of compensation is not unlimited.
Most progress on defining the mechanistic basis for the effects of alcohol on alveolar barrier function have been through studies of rodents fed the isocaloric Lieber DeCarli alcohol diet that carefully controls for nutrition and thereby can “isolate” the independent effects of alcohol ingestion [21]. Studies in the intact animal have demonstrated that rats on a chronic alcohol diet have enhanced permeability to small molecules (e.g., inulin) as well as to proteins as large as serum albumin [22]. Moreover, their ability to resist a saline challenge is impaired. Understanding this at a molecular level has benefited from the observation that the “alcoholic lung” phenotype of alveolar epithelial cells isolated from alcohol-fed rats persists in culture. In other words, alveolar epithelial cells isolated from alcohol-fed rats and grown in monolayers in culture remain leakier than comparably isolated and cultured cells from control-fed rats [22]. The basis for this is not fully understood, but it is likely that alveolar epithelial cells differentiate in response to chronic stress in the alcoholic lung and that this results in epigenetic modification to genomic DNA that modifies the behavior of these cells [23]. Epigenetics is a nascent field in alcohol research and is only beginning to be explored [24, 25]. Given the persistence of the alcoholic lung phenotype, alveolar epithelial cells from alcohol-fed rats provide a valuable model system to study their pathophysiology.
Tight Junctions and Alveolar Barrier Function
The bulk of the alveolar barrier consists of the cells themselves. The remaining barrier function is due to a series of structures at cell–cell interfaces called tight junctions. Tight junctions serve as a sealing point between polarized epithelial cells and denote the boundary between the apical and basolateral plasma membrane domains (Fig. 8.2). This segregation and sealing function formed by the tight junctions provide two important physiological functions of the epithelium: (1) a physical barrier to small molecules and (2) a series of paracellular ion channels that enable ion diffusion between cells [26].
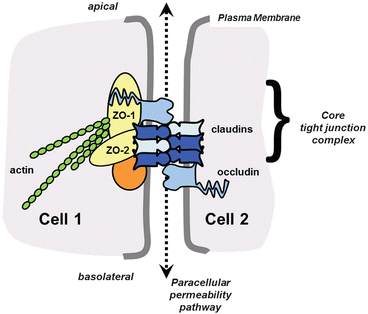
Fig. 8.2
Core tight junction protein complex. Shown are the major functional components of tight junctions known to directly interact. These include transmembrane proteins (claudins, occludin), scaffold proteins (ZO-1, ZO-2), and the actin cytoskeleton. Head-to-head interactions between claudins on adjacent cells form the basis for paracellular channels which restrict permeability. Homotypic interactions (between the same type of claudin) and heterotypic interactions (between different types of claudins) are depicted. The likely presence of additional scaffold proteins is represented by the orange circle. Not shown are immunoglobulin-fold transmembrane proteins, which associate with tight junctions and also regulate claudin expression and tight junction permeability, such as Junction Adhesion Molecule A (JAM-A) [116]. Adapted from [26]
Tight junctions are formed by a complex that includes transmembrane proteins, cytosolic scaffold proteins, and cytoskeletal proteins. Tight junction permeability is primarily mediated by proteins known as claudins that span the membrane bilayer four times and have both the N- and C-terminal domains oriented inside towards the cytosol. The C terminus of claudins interacts with cytosolic scaffold proteins, most notably zonula occludens-1 (ZO-1) and ZO-2 [27]. ZO-1 and ZO-2 in turn link claudins to the actin cytoskeleton. While almost all claudins interact with ZO-1 and ZO-2, they differ in the extent of actin association, which correlates with incorporation into functional tight junctions. Other transmembrane proteins also regulate tight junction assembly and stability, particularly occludin and tricellulin [28]. Roles for these proteins in the alveolar epithelium are continuing to be elucidated. However, occludin and ZO-1 expression are both down-regulated by the alveolar epithelium in response to alcohol, consistent with the concept that an early and primary defect in the alcoholic lung is impaired alveolar epithelial barrier function [29, 30].
There are over a dozen claudin isoforms expressed by the alveolus; the nine most prevalent are summarized in Table 8.1. Of these, 97 % of claudin mRNA in both type I and type II cells encodes for claudin-3, claudin-4, and claudin-18 [31]. However, type II cells express significantly more claudin-3 than is expressed by type I cells. This difference has functional ramifications, since claudin-3 expression increases alveolar tight junction permeability whereas claudin-4 makes tight junctions less permeable (Fig. 8.3) [32]. One consequence of this difference is that type II–type I cell interfaces are enriched for claudin-3 and are therefore likely to be more permeable than type I–type I cell interfaces. Functionally, the unique permeability of type II–type I cell junctions might provide a paracellular pathway for ion and fluid diffusion that counterbalances ion flux due to channel activity (see below).
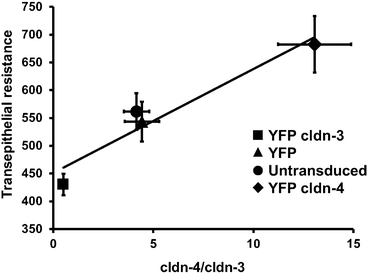
Table 8.1
Alveolar epithelial claudin expression
Human | Rat | Mouse | |||
|---|---|---|---|---|---|
Claudin-3 | RPa | P | RPb | RPb | P |
Claudin-4 | RP | P | RP | RP | P |
Claudin-18 | RP | P | RP | RP | P |
Claudin-5 | RP | P | RP | RP | P |
Claudin-7 | RP | RP | RP | P | |
Claudin-10b | R | R | R | P | |
Claudin-12 | R | RP | |||
Claudin-15 | R | RP | |||
Claudin-19 | R | R | |||
Fig. 8.3
Differential effect of increasing cldn-3 or cldn-4 on alveolar epithelial cell barrier function. Model type I alveolar epithelial cells transduced with YFP-cldn-3 (filled square), YFP-cldn-4 (filled diamond), YFP-control virus (filled circle), or untransfected controls (filled triangle) were assessed for the effect of altering claudin expression on barrier function, as determined using transepithelial resistance (TER; Ohm × cm2) (Y-axis). The expression ratio cldn-4/cldn-3 was determined by immunoblot (X-axis) demonstrating that there was a linear relationship between cldn-4/cldn-3 ratio and TER (r 2 = 0.93). Cells expressing increased cldn-3 had significantly lower TER than either control cells or cells expressing increased cldn-4 (p <0.05). Increasing cldn-4 also significantly increased barrier function (p < 0.05). Adapted from [37]
Critically, claudin-4 is up-regulated during ventilator-induced lung injury. Since claudin-4 expression correlates well with human lung fluid clearance and is inversely related to the severity of the Acute Respiratory Distress Syndrome (ARDS), this underscores a protective effect of claudin-4 in lung function [33, 34]. Claudin-18 has a longer C-terminal domain and is more tightly associated with the cytoskeleton than claudin-3 or claudin-4, most likely due to increased interactions with tight junction scaffold proteins [32]. Claudin-18 is decreased in response to inflammation [35], consistent with impaired barrier function in ARDS.
As mentioned above, alcohol impairs alveolar barrier function by increasing tight junction permeability. In part this is due to decreases in claudin expression [36]. Perhaps more critically, the decrease in alveolar barrier function due to alcohol is accompanied by a significant shift in tight junction morphology as determined by immunofluorescence microscopy (Fig. 8.4). In normal cells, claudins are predominantly localized to cell–cell contact sites. By contrast, cells impaired by alcohol show significant morphological disruption of claudin localization, including strand breaks and increased intracellular labeling. Intriguingly, this correlates with an increase in expression of claudin-5, which is normally expressed at low levels in the alveolus [36, 37]. These effects of alcohol on claudin-5 and epithelial barrier function are consistent with the observations that increased claudin-5 correlates with impaired lung epithelial barrier function in cells treated with methanandamide or in cells directly transfected with claudin-5 [38, 39]. However, whether there is a direct mechanistic link between claudin-5 and disruption of tight junction assembly and function in alveolar epithelium remains to be determined.
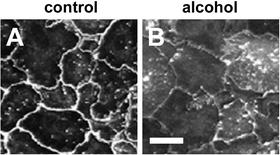
Fig. 8.4
Alcohol impairs assembly of claudins into tight junctions. Model type I alveolar epithelial monolayers were derived from primary cells isolated from either control (a) or alcohol-fed (b) rats which were cultured for 6 days and then immunolabeled for cldn-7. In contrast with control alveolar epithelial cells, where cldn-7 prominently localized to sites of cell–cell contact (a), cells isolated from alcohol-fed rats had impaired claudin assembly (b) which correlated with impaired barrier function. Bar—10 μm. Adapted from [37]
Ion Channels and Alveolar Fluid Clearance
The primary mode of fluid efflux from airspaces is promoted by ion flux through alveolar epithelial cells that sets up an electrochemical gradient that drives water from airspaces into tissues. The majority of ion flux is due to sodium transport. On the apical surface, sodium transport into cells is mediated by epithelial sodium channels (ENaC), which are the rate-limiting step in alveolar sodium transport [40]. Transport out of cells on the basolateral surface is by Na,K-ATPases that actively pump sodium out of the cell and thereby provide the gradient that promotes transcellular passage of sodium from the alveolar space into the interstitium. Both type I and type II cells express sodium channels, as well as other ion transporters, that contribute to maintenance of lung fluid balance.
In contrast to the effect of alcohol on barrier function, alcohol increases net flux of sodium by having a direct effect on both parts of the sodium transport pathway. This was confirmed by examining the expression of lung Na,K-ATPase in rats fed the Lieber-DeCarli diet [41]. Na,K-ATPase is composed of several subunits, all of which were up-regulated in the alcoholic lung. When treated with the Na,K-ATPase inhibitor ouabain, lungs from alcohol-fed rats developed more edema than control lungs, indicating that increased Na,K-ATPase compensates for other deficits in maintenance of lung fluid balance induced by alcohol.
In parallel to this increase in Na,K-ATPase expression, ENaC is also up-regulated in the alcoholic lung [42]. Critically, ENaC activity was also increased by direct oxidation mediated through NADPH oxidase activity. This is in line with other studies demonstrating that reactive oxygen species have the capacity to increase ENaC and lung fluid clearance [43, 44]. Together, increased ENaC and Na,K-ATPase in the alcoholic lung provide a pathway for up-regulated fluid clearance that can compensate for deficits in barrier function induced by dietary alcohol. Studies to date have focused on sodium transport; chloride transport must also increase in order to retain electronic neutrality across the alveolar epithelium. Whether this occurs via up-regulation of chloride channels or by chloride transport via the paracellular route remains an open question [40]. Taken together, these observations explained an initial paradox when the phenotype of the alcoholic lung was first being examined experimentally. Specifically, if chronic alcohol ingestion causes such significant defects in alveolar epithelial permeability, why don’t alcoholics have pulmonary edema at baseline? The marked up-regulation of active fluid transport mechanisms in the alcoholic lung appear to compensate for the increased paracellular permeability and maintain a relatively “dry” airspace. However, this new equilibrium is far more susceptible to the stresses imposed by acute inflammation such as pneumonia or sepsis, and the alcoholic lung is far more prone to alveolar flooding with proteins and fluids during critical illness.
Therefore, in the otherwise healthy alcoholic, barrier function and fluid efflux are counterbalanced to maintain a non-edematous lung. However, if alveolar barrier function is further impaired due to a second hit, e.g., sepsis or barotrauma, the lung requires an additional increase in sodium transporters in order to prevent alveolar edema. ENaC in the alcoholic lung does have some additional capacity for further up-regulation (e.g., in acute response to endotoxin [42]). However, it is clear that lungs of injured alcohol-fed animals are more susceptible to endotoxin and sepsis induced lung edema than non-alcohol-fed controls [2, 18]. This suggests that in the alcoholic lung, fluid clearance saturates at a level insufficient to compensate for barrier dysfunction which, in turn, exacerbates the severity of ARDS. Therefore, any treatment modality designed to prevent alcohol-associated ARDS must address alveolar tight junctions, since it is not likely to be possible to increase fluid clearance enough to prevent alveolar flooding.
Intercellular Communication
Gap Junctions
Gap junctions provide a means for intracellular communication by forming channels that connect the cytosols of adjacent cells in a tissue (Fig. 8.5). This connection enables the direct diffusion of small cytoplasmic molecules, ions, and water. Metabolites including adenosine triphosphate (ATP) and glutathione also can move through gap junction channels. Several functions have been ascribed to gap junctions including metabolite sharing, electrical coupling, cell growth control, and regulation of cell migration.
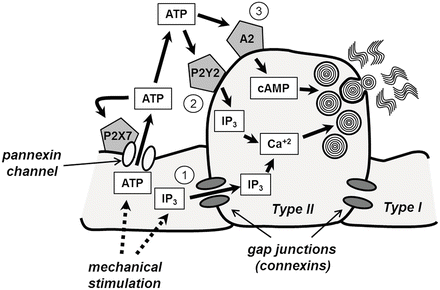
Fig. 8.5
Intercellular communication pathways between alveolar epithelial cells which control surfactant secretion. Gap junctional transmission (1) of either calcium or inositol trisphosphate (IP3) generates increased cytosolic calcium in type II cells, which, in turn, could stimulate surfactant secretion either through protein kinase C activation or through a direct effect of calcium on lamellar body fusion. Increased cytosolic calcium can also be mediated through secretion of ATP through P2X7 receptor-mediated pannexin channels (2) acting on P2Y2 purinergic receptors. Protein kinase A activation through stimulation of A2 purinergic receptors by ATP can also occur (3). Adapted from [46]
The proteins that form gap junctions are known as connexins. In the alveoli, connexin-43 (Cx43) is the major connexin demonstrated to have functional roles [45–49]. Other connexins expressed by the alveolar epithelium include Cx26, Cx32, Cx30.3, Cx40, and Cx46. Specific roles for these other connexins in the alveolar epithelium have not been elucidated. Among these, Cx32 is restricted to type II cells and is absent from type I cells [50, 51]. Moreover, type I cells cannot form functional gap junctions with cells expressing only Cx32; however, type II cells readily communicate through Cx32 gap junction channels [51]. Although type I cells can form functional heterocellular gap junctions with type II cells via Cx43, roles for Cx32 in regulating alveolar signaling remain unknown at present. One possibility is that Cx32 provides a “priority channel” where type II cells can specifically communicate with each other, but this has yet to be determined.
Alcohol treatment of cells in vitro inhibits gap junctional communication [52–55]. Specifically, alcohol significantly inhibits expression of the major lung connexin, Cx43 [52, 53], but has less effect on other connexins [52, 54]. A direct mechanism for the effect of alcohol on gap junctions is by partitioning into membranes and changing the connexin microenvironment [56]. Hormones induced by chronic alcohol ingestion can also have deleterious effects on alveolar gap junctions. Most notably, TGF-β inhibits alveolar Cx43 expression [55]. Moreover, glutathione depletion and increased oxidant stress induced by chronic alcohol exposure also decrease gap junctional communication [57, 58]. Although a reduction in gap junctions in response to oxidant stress reduces intercellular signaling, it can be beneficial since it decreases the transfer of toxic agents and thus minimizes tissue damage [59, 60]. However, this is at the expense of promoting the transfer of protective agents including glutathione.
Recently, a novel mode of Cx43-dependent interaction in the alveolus was demonstrated by showing that bone-marrow-derived stromal cells (BMSCs) instilled into injured lungs formed functional gap junctions with type II cells in vivo [61]. This interaction was particularly critical for preventing cell injury due to insufficient mitochondrial function. In part, this could be due to direct transfer of metabolites from healthy BMSCs to damaged type II cells. More provocatively, evidence was provided showing direct mitochondrial transfer from the BMSCs to the type II cells, which also appeared to require functional gap junctions. Gap junction permeability is too limited to enable direct movement of mitochondria through channels themselves. Instead, mitochondrial transfer was due to microvesicles that were taken up by type II cells. It is plausible that this gap junction plaque endocytosis could enable mitochondrial transfer since, during this process, the plasma membrane and a small amount of cytosol are taken up by the neighboring cell [62]. Regardless of the specific mechanism, if a mitochondrial transfer pathway is critical for ameliorating lung injury then it is likely to be adversely affected by alcohol, which impairs both gap junctional communication (see above) and mitochondrial function [63].
Gap junctions play an important role in regulating the secretion of pulmonary surfactant produced by type II alveolar epithelial cells. Mechanical distension of type I cells in the alveolus occurs during a physical stretch, for example a deep breath or assisted ventilation, which then initiates a transient increase in cytosolic calcium through stretch-activated channels. The calcium is then transmitted through gap junctions from type I cells to type II cells where it stimulates fusion of lamellar bodies with the plasma membranes to release surfactant [64–66].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


