Fig. 7.1
Beneficial and detrimental effects of alcohol drinking on cardiovascular risk factors and cardiovascular events. Each effect of alcohol depends on the degree of alcohol consumption. CVD cardiovascular diseases, SCD sudden cardiac death
Among alcoholic beverages, additional benefits for cardiovascular protection are expected in red wine because it contains polyphenols, which exert cardiovascular protective actions represented by antioxidative action (Rodrigo et al. 2011). However, whether red wine is superior to other alcoholic drinks or not is still debatable because there is a possibility of confounding by socioeconomic factors such as a healthier lifestyle of wine drinkers compared to that of beer and spirit drinkers (van de Wiel and de Lange 2008). Incidental heavy or binge drinking (generally defined as five or more drinks on an occasion) is known to increase cardiovascular events, and thus the pattern of drinking is suggested to be more important than the kind of alcoholic beverage (van de Wiel and de Lange 2008). Wine consumption with a meal has been shown to suppress blood level of postprandial lipid hydroperoxides (Ursini et al. 1998), and alcohol intake with a meal may have beneficial effects on metabolism of alcohol by its retarded absorption. Therefore, regular light-to-moderate drinking (no more than one drink per day for women and two drinks per day for men) with a meal is the way to go for drinkers. On the other hand, the use of alcohol as a cardioprotective strategy is not recommended for abstainers because of no proof of causality and no way to predict individual possibility of alcohol abuse in the future.
7.2 Alcohol and Blood Lipids
An abnormal blood cholesterol profile, such as high LDL cholesterol and low HDL cholesterol, is the most important risk factor for coronary arterial disease. Habitual alcohol drinking results in elevation of HDL cholesterol and reduction of LDL cholesterol (Castelli et al. 1977; Langer et al. 1992; Gaziano et al. 1993). About half of the protection against coronary heart disease afforded by moderate alcohol consumption has been reported to be mediated by an increase in HDL cholesterol, and an additional 18 % of this protection has been shown to be attributable to a decrease in LDL cholesterol (Langer et al. 1992). Alcohol induces elevation of HDL cholesterol, especially its subfractions of HDL2 and HDL3 (Gaziano et al. 1993). In fact, HDL2 sampled from heavy drinkers has been demonstrated to enhance cholesterol efflux from cultured macrophages in vitro (Mäkelä et al. 2008). Non-HDL cholesterol, calculated as the difference between total cholesterol and HDL cholesterol, contains particles of all atherogenic apolipoprotein B-containing lipoproteins, including very-low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), LDL, and lipoprotein(a), and has been reported to be superior to LDL cholesterol in predicting cardiovascular events (Cui et al. 2001). Non-HDL cholesterol has also been shown to be lower in drinkers than in nondrinkers (Wakabayashi and Groschner 2009).
As mechanisms for the effects of alcohol on lipid metabolism, habitual alcohol drinking has been shown to increase lipoprotein lipase (LPL) activity and decrease cholesteryl ester transfer protein (CETP) activity, resulting in elevation of HDL cholesterol and reduction of LDL cholesterol (Savolainen et al. 1990; Hannuksela et al. 2004). CETP mediates transfer of cholesteryl esters from HDL to LDL and VLDL particles. Alcohol itself or its metabolite, acetaldehyde, has been suggested to cause defective sialylation and inhibit glycosylation of CETP, which is necessary for binding of CETP to lipoproteins, in alcohol drinkers (Liinamaa et al. 2006). Polymorphism of the CETP gene (TaqIB) is known to influence HDL cholesterol level and risk of coronary artery disease (Boekholdt et al. 2005). The TaqIB SNP has been reported to modulate effects of alcohol on level of HDL cholesterol and risk of coronary artery disease: Individuals carrying the TaqIB2 allele showed stronger associations of alcohol intake with elevation of HDL cholesterol and reduction of the risk of coronary artery disease than did individuals carrying the TaqIB1 allele (Fumeron et al. 1995). Regarding polymorphism of LPL, S447X polymorphism has been reported to modulate the association between alcohol drinking and plasma HDL cholesterol level in women (Lee et al. 2004).
A meta-analysis study using 35 data records has shown that triglyceride concentrations increased by 0.19 mg/dl/g of alcohol consumed a day and that this association was stronger in men than in women and slightly stronger in beer drinkers than in wine drinkers (Rimm et al. 1999). Excessive alcohol drinking causes hypertriglyceridemia in both postprandial and fasting states, which may be mainly due to an increase in the synthesis of large VLDL particles in the liver, and an acute inhibitory effect of alcohol on LPL activity is also involved in postprandial hypertriglyceridemia in drinkers (Van de Wiel 2012). On the other hand, a well-controlled intervention study has demonstrated that consumption of alcohol (15 and 30 g/day) dose dependently reduced triglyceride levels in healthy postmenopausal women (Davies et al. 2002). Therefore, the relationship between alcohol and triglycerides may be greatly modified by diet and other lifestyle factors.
7.3 Alcohol and Blood Coagulation-Fibrinolysis Balance
Thrombus formation on the arterial wall is an important etiology for progression of atherosclerosis. Therefore, a disorder of blood coagulation-fibrinolysis balance determined by activities of platelets, coagulation factors, and fibrinolysis factors is a major risk factor for atherosclerotic disease. Inhibitory effects of alcohol on platelet function and blood coagulation factors and augmenting effects of alcohol on the blood fibrinolysis pathway partly explain the inverse association between light-to-moderate alcohol consumption and the risk for ischemic types of atherosclerotic diseases such as ischemic heart disease, cerebral infarction, and peripheral arterial disease. On the other hand, hemorrhagic stroke is induced by heavy drinking through potentiation of fibrinolytic function by alcohol.
Alcohol inhibits platelet activation in vivo and in vitro in response to diverse agonists such as collagen, adenosine diphosphate (ADP), thrombin, and platelet-activating factor (PAF). Ethanol inhibits the formation of arachidonic acid and thromboxane A2, while free arachidonic acid-induced platelet aggregation was not affected by ethanol (Rubin 1999). Direct inhibition of membrane-associated phospholipase A2 by ethanol was also demonstrated (Stubbs and Rubin 1992). Therefore, the major inhibitory focus of ethanol is thought to be phospholipase A2. A physiological agonist, such as thrombin, induces platelet aggregation via phospholipase C-mediated phosphoinositide turnover, resulting in production of inositol trisphosphate and diacylglycerol. The former induces release of Ca2+ from its intraplatelet storage pools and triggers transmembraneous Ca2+ entry, called capacitative Ca2+ entry (CCE) or store-operated Ca2+ entry (SOCE), in which canonical transient receptor potential channel (TRPC) 1/4/5 is involved. The latter product induces transmembraneous Ca2+ entry through activating TRPC 3/6/7. Ethanol has been demonstrated to inhibit agonist-independent CCE induced by a Ca2+-ATPase inhibitor, thapsigargin (Wakabayashi and Marumo 2002), while Ca2+ entry induced by a diacylglycerol mimic, 1-oleoyl-2-acetyl-sn-glycerol (OAG), was not inhibited by ethanol (Marumo and Wakabayashi 2010). Therefore, ethanol is thought to selectively inhibit TRPC 1/4/5. As another inhibitory focus of ethanol, ADP- and thrombin-induced p-selectin expression in platelets has been shown to be attenuated by ethanol, and this inhibitory effect of ethanol is suggested to be independent of the effect of ethanol on arachidonate metabolism, because aspirin treatment did not attenuate this effect (Nguyen et al. 1998; Mukamal et al. 2005).
Elevated fibrinogen level in blood is known to be an independent cardiovascular risk factor, although it is still under debate whether fibrinogen is just a marker of cardiovascular risk or whether lowering circulating levels of fibrinogen will result in a significant decrease in clinically relevant endpoints (Kakafika et al. 2007). An inverse association between alcohol consumption and fibrinogen level has been shown in many epidemiological studies. A 0.78 % decrease in fibrinogen concentration with each 10 g/day increase in alcohol intake has been indicated by results of a prospective study (Meade et al. 1987). An experimental study demonstrated that blood fibrinogen concentration was lowered by daily ethanol administration in rats (5 % ethanol v/v in drinking water for 4 weeks) and that fibrinogen production and mRNA expression of fibrinogen in lined hepatoma cells were diminished by exposure of the cells to ethanol at 10 and 20 mM (Wang et al. 1999). In addition to fibrinogen level, levels of von Willebrand factor and factor VII have been shown to be lower in light-to-moderate alcohol drinkers than in nondrinkers (Mukamal et al. 2001).
The results of previous epidemiological studies concerning relationships of alcohol drinking with fibrinolytic factors are controversial. Positive associations of alcohol intake with tissue plasminogen activator (tPA) antigen and activity and with urine plasminogen activator (uPA) activity were demonstrated (Sumi et al. 1988; Ridker et al. 1994; Mukamal et al. 2001), while plasminogen activator inhibitor-1 (PAI-1) antigen and activity were reportedly increased by acute alcohol drinking (Mukamal et al. 2001; van de Wiel et al. 2001). There were reports showing no relationships of regular alcohol drinking with tPA antigen and activity levels and PAI-1 antigen and activity levels (van Golde et al. 2002; Volpato et al. 2004). In cultured human monocytes, fibrinolytic activity and mRNA expression of tPA and uPA were increased by incubation with ethanol (0.1 % v/v) (Tabengwa et al. 2002). Ethanol in vitro also increased tPA and uPA antigen and their mRNA expression in cultured human vascular endothelial cells, while PAI-1 antigen and its mRNA expression were decreased by ethanol in vitro (Booyse et al. 1999).
7.4 Alcohol-Induced Hypertension
Since the 1970s, many longitudinal and cross-sectional studies have demonstrated that chronic alcohol consumption is a risk factor for hypertension independent of the type of alcoholic beverage, adiposity, education, smoking, salt intake, and several other traits (Klatsky 1996). A study analyzing WHO Global Burden of Disease has shown that 16 % of all hypertensive diseases were attributed to alcohol (Rehm et al. 2003). A variety of mechanisms for the relationship between alcohol and blood pressure have been suggested by previous studies using acute and chronic experiments. The mechanisms include increased levels of catecholamines, renin, and cortisol; sympathetic nervous system effect; increased peripheral vascular muscle tone; abnormality in calcium transport; erythrocyte membrane changes; insulin resistance; and magnesium depletion (Klatsky 2004). However, no convincing conclusion with respect to the exact mechanism for alcohol-induced hypertension has been obtained. According to a systemic review on the relationship between alcohol and blood pressure including studies using ambulatory or home blood pressure monitoring, alcohol shows two opposite effects, an early effect (in the hours after drinking) leading to a reduction of blood pressure and a later effect (next day) leading to an elevation of blood pressure (McFadden et al. 2005). Because habitual drinkers experience repeated alcohol withdrawal, adrenergic discharge following withdrawal from alcohol intake has been suggested to be an important mechanism for alcohol-induced hypertension (Potter et al. 1983; Kawano 2010). As the mechanism for the acute depressing effect of alcohol on blood pressure, several actions of acetaldehyde, such as direct vasodilating action (Altura and Altura 1982), release of prostacyclin (Guivernau et al. 1987), and activation of glandular kallikreins (Hatake et al. 1990), have been proposed. Since changes in average 24-h blood pressure after drinking alcohol are smaller than alcohol-induced changes in blood pressure at casual office- or clinic-based measurements, a possibility of overestimation of the hypertensive effect of alcohol has been pointed out (Kawano 2010).
7.5 Alcohol and Cardiac Arrhythmias
High risk for cardiac rhythm disorders, particularly atrial fibrillation, by heavy alcohol drinking on weekends or holidays is known as the “holiday heart syndrome” (Ettinger et al. 1978). Meta-analysis studies have demonstrated a dose-dependent relationship between alcohol drinking and risk of atrial fibrillation: The risk was estimated to be increasing 1.5-fold by the highest alcohol intake compared to that by the lowest alcohol intake or no alcohol intake, while there may be no difference in the risk between abstainers and moderate drinkers (Samokhvalov et al. 2010; Kodama et al. 2011). Decrease in calcium currents and shortening of action potential duration by alcohol were demonstrated in in vitro studies but were not reproduced in studies using intact animal models, and results regarding electrophysiological effects of alcohol in humans were controversial (Balbão et al. 2009). Therefore, at present, alcohol seems not to exert direct myocardial toxicity inducing atrial fibrillation and is speculated to cause susceptibility to induction of atrial fibrillation through increasing levels of circulating catecholamines, inducing oxidative stress, and releasing plasma free fatty acids in individuals with structural heart diseases or in long-term alcohol drinkers with subclinical cardiac abnormalities (Balbão et al. 2009).
Heavy drinking is also associated with increased risk of ventricular tachyarrhythmias and sudden cardiac death (SCD). Potential mechanisms that could account for these associations are related to multiple physiological aberrancies in chronic alcohol abuse, including increased QT interval leading to ventricular tachyarrhythmias, proarrhythmic electrolyte abnormalities such as hypomagnesemia and hypokalemia, sympathoadrenal stimulation, and reduced baroreflex sensitivity (Lucas et al. 2005; George and Figueredo 2010). In contrast, a prospective study has shown reduced risk of SCD in light alcohol drinkers (two to six drinks per week) compared with that in subjects who rarely or never drank alcohol and compared with heavy drinkers (six or more drinks per day) and binge drinkers (Albert et al. 1999). This inverse association between modest alcohol consumption and risk of SCD may be attributable to reduced risk of coronary artery disease in light-to-moderate alcohol drinkers.
7.6 Alcohol and Cardiomyopathy
Alcoholic cardiomyopathy, an important etiology of mortality in chronic alcoholics, is observed in those excessively consuming alcohol such as greater than 100 and 80 g of ethanol per day in men and women, respectively, for longer than 10 years in the absence of coronary artery disease and nutritional disorders (Urbano-Marquez et al. 1989). Thus, women are more sensitive to toxic effects of alcohol on the myocardium than are men. There may also be a genetic involvement in vulnerability to alcoholic cardiomyopathy: Alcoholics with DD polymorphism of angiotensin-converting enzyme (ACE) showed a 16-fold higher risk of cardiomyopathy than did those with other ACE genotypes (Fernández-Solà et al. 2002). Left ventricular diastolic dysfunction has been proposed to be the earliest sign of subclinical alcoholic cardiomyopathy. After subclinical alcoholic cardiomyopathy, patients continuing to drink alcohol heavily suffer from congestive heart failure and have a higher incidence of arrhythmias and sudden death.
Macro- and microscopic findings of alcoholic cardiomyopathy are indistinguishable from those of other dilated cardiomyopathy. In vitro and ex vivo experiments revealed that long-term alcohol consumption induced histological and cellular abnormalities such as myocyte loss, intracellular organelle dysfunction, reduction of contractile proteins, and impairment of calcium homeostasis (Laonigro et al. 2009). Alcohol has been shown to have apoptotic effects on cardiac myocytes and increase expression of pro-apoptotic proteins such as Bax and Bcl-2 with degrees similar to the effects of long-standing hypertension (Fernández-Solà et al. 2006). Increase in myocardial lipid peroxidation and protein oxidation and decrease in glutathione content have been demonstrated in alcohol-intoxicated animal models, and thus reactive oxygen species (ROS), which are produced from ethanol through cytochrome P450 2E1 after heavy drinking, play an important role in alcohol-induced cardiomyopathy. The renin-angiotensin system (RAS) is activated by prolonged alcohol consumption (Wright et al. 1986). Alcoholic cardiomyopathy has also been proposed to be caused by alcohol-generated nitrative stress through activation of protein kinase C (PKC)-β1-dependent expression of NOX (nicotinamide adenine dinucleotide phosphate oxidase) by interaction of stimulation of angiotensin II with its type 1 receptor (Tan et al. 2012). A mass spectrometry-based proteomic analysis demonstrated that chronic ethanol administration caused reduction of myofibrillar, sarcoplasmic, membrane-associated, and mitochondrial proteins due to fundamental alteration of cardiac muscle (Fogle et al. 2010). Chronic alcohol consumption has been shown to impair myocardial protein synthesis through RNA translation following reduction of active eIF4E (eukaryotic translation initiation factor 4E) and p70 ribosomal S6 phosphorylation (Vary et al. 2001). Ethanol is known to impair cardiac excitation-contraction coupling, and abnormalities in calcium homeostasis, such as impairment of cytosolic Ca2+ transients, decrease in myofilament Ca2+ sensitivity, and upregulation of L-type Ca2+ channels, have been demonstrated in alcohol-intoxicated rat hearts (Laonigro et al. 2009). The above-proposed mechanisms for alcohol-induced cardiomyopathy are summarized in Fig. 7.2.
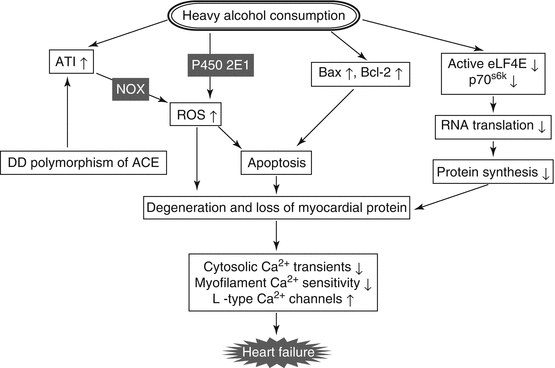
Fig. 7.2
Proposed mechanisms for alcohol-induced cardiomyopathy. AT1 angiotensin type 1 receptor, ACE angiotensin-converting enzyme, NOX nicotinamide adenine dinucleotide phosphate oxidase, p70S6K p70 ribosomal S6 kinase, ROS reactive oxygen species
7.7 Alcohol and Hyperuricemia
In addition to relations to established cardiovascular risk factors such as gender, age, obesity, and blood pressure, elevated serum uric acid level is an independent risk factor for cardiovascular disease (Feig et al. 2008). However, it has not yet been determined whether lowering uric acid levels would be of clinical benefit for the prevention of cardiovascular disease (Feig et al. 2008). In vivo and in vitro findings suggest that uric acid contributes to endothelial dysfunction and has proinflammatory and proliferative effects on vascular smooth muscle cells (Kanellis and Kang 2005). In a US prospective study (NHANES III), serum uric acid level tended to increase with an increase in frequency of drinking and was highest in habitual beer drinkers among various alcohol beverage drinkers, while there was an inverse relationship between drinking frequency and serum uric acid level in habitual wine drinkers (Choi and Curhan 2004). In addition to intake of purine body contained in alcohol beverages, particularly in beer, alcohol drinking increases uric acid level by reducing renal excretion of uric acid due to elevation of blood lactate and by increasing uric acid production due to enhanced turnover of adenine nucleotides following consumption of ATP in the liver (Yamamoto et al. 2005). On the other hand, acute elevation of uric acid has recently been reported to increase antioxidant capacity and to be a protective factor when drinking red wine (Modun et al. 2008).
7.8 Red Wine Polyphenols and Cardiovascular Disease
Polyphenols are mainly responsible for reduced risk of cardiovascular diseases by red wine, which is known as French paradox and Mediterranean diet. Polyphenols in red wine, most of which are derived from the skin and seeds of grapes, possess antioxidant and free radical-scavenging properties and show beneficial effects through acting on signaling pathways of platelets and vascular cells such as endothelial and smooth muscle cells. In addition, lipid- and lipoprotein-lowering actions of polyphenols are also involved in reduction of cardiovascular disease risk by red wine and its polyphenol constituents. Major wine-containing polyphenols are stilbenes, flavan-3-ols, flavonols, anthocyanins, hydroxybenzoic acid, procyanidins, and hydroxycinnamic acids, and they are largely grouped as flavonoids and nonflavonoids (Smirnoff 2005). Well-known flavonoids in red wine are flavonols (e.g., quercetin) and flavan-3-ols (e.g., tannins and catechin), while resveratrol is the best known nonflavonoid.
Phenolic compounds in red wine, but not its alcohol component, have antioxidative characteristics including scavenging of free radicals, chelation of metals, sparing of vitamin E and carotenoids, and increasing serum paraoxonase activity (Fuhrman and Aviram 2001). Increase in blood antioxidant activity following red wine consumption reduces LDL sensitivity to peroxidation and thereby suppresses foam cell formation and early atherosclerotic plaque development. Figure 7.3 shows various mechanisms explained below for suppression of atherosclerosis by polyphenols in alcohol beverages.
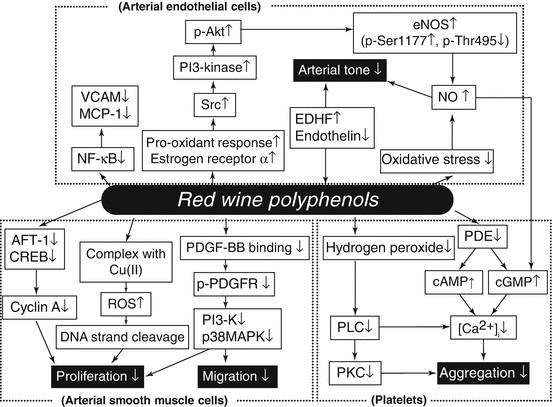
Fig. 7.3
Proposed mechanisms for suppression of atherosclerosis by polyphenols contained in alcohol beverages. ATF-1 activating transcription factor-1, CREB cAMP-responsive element-binding protein, eNOS endothelial nitric oxide synthase, EDHF endothelium-derived hyperpolarizing factor, MCP-1 monocyte chemotactic protein-1, NFκB nuclear factor κB, NO nitric oxide, PDE phosphodiesterase, PDGF platelet-derived growth factor, p-PDGFR phosphorylated platelet-derived growth factor receptor, PI3-K phosphoinositide 3-kinase, PLC phospholipase C, PKC protein kinase C, ROS reactive oxygen species, VCAM vascular cell adhesion molecule
Red wine has been demonstrated to inhibit agonist-induced platelet aggregation and thromboxane A2 formation by in vitro and ex vivo experiments (Pace-Asciak et al. 1996). Raising levels of cAMP and cGMP in platelets due to decreased activities of cAMP and cGMP phosphodiesterases and subsequent decrease in cytosolic calcium levels have been shown to be responsible for polyphenol-induced inhibition of platelet function (Soleas et al. 1997). Red wine polyphenols also decreased platelet production of hydrogen peroxide and inhibited the phospholipase C pathway (Pignatelli et al. 2000). Nitric oxide (NO) inhibits platelet aggregation through augmenting cGMP guanylate cyclase, resulting in an increase in cGMP level in platelets. Grape juice has been shown to attenuate platelet aggregation, increase platelet-derived NO release, and decrease superoxide production in platelets (Freedman et al. 2001).
The vascular endothelium plays obligatory roles in preserving blood flow and keeping a good coagulation-fibrinolysis balance. For these purposes, NO is a key substance produced in and released from endothelial cells. Red wine polyphenols are known to enhance endothelial NO production and NO-mediated vasodilation (Fitzpatrick et al. 1993). Oxidative stress reduces vascular bioavailability of NO, causing an increase in vascular tone. Polyphenols have been shown to reduce vascular oxidative stress through reducing expression and activity of NADPH oxidase and increasing antioxidant enzymes such as catalase (Ying et al. 2003). Several mechanisms for enhancement of endothelial NO synthase (eNOS) by polyphenols have been proposed: Src-dependent activation of the PI3-kinase pathways in endothelial cells is induced by prooxidant response to polyphenols, resulting in Akt phosphorylation, which increases activity of eNOS via phosphorylation of Ser1177, an activator site of eNOS, while Thr495, an inhibitor site of eNOS, is dephosphorylated by red wine polyphenols (Ndiaye et al. 2005; Anselm et al. 2007). Estrogen receptor α has been suggested to be a pivotal receptor of red wine polyphenols, particularly delphinidin, involved in Src-dependent activation of eNOS expression (Chalopin et al. 2010). As other mechanisms for the vasodilating activity of red wine polyphenols, inhibition of endothelin-1 synthesis (Corder et al. 2001) and induction of EDHF (endothelium-derived hyperpolarizing factor)-mediated vasorelaxation (Ndiaye et al. 2003) have been reported in several vessels.
Endothelial expression of an adhesion molecule, VCAM-1 (vascular cell adhesion molecule-1), which is activated through the NFκB (nuclear factor κB) pathway, is increased at atherosclerotic lesions. Red wine inhibits NFκB activation, resulting in downregulation of VCAM-1 (Carluccio et al. 2003). Red wine also inhibits endothelial expression of MCP-1 (monocyte chemotactic protein-1), resulting in attenuated monocyte migration into the subendothelial lesion (Feng et al. 1999).
Vascular smooth muscle cell proliferation and migration, which are important components of atherogenesis, are inhibited by polyphenols. Several mechanisms for the antiproliferative action of red wine have been proposed: Gene expression of cyclin A, a cell-cycle regulator functioning at two stages of the cell cycle, at the G1-S transition and again at the G2-M transition, is downregulated by polyphenols through decreasing transcription factors such as AFT-1 (activating transcription factor-1) and CREB (cAMP-responsive element-binding protein) (Iijima et al. 2000). Platelet-derived growth factor (PDGF)-BB, released from platelets, endothelial cells, and smooth muscle cells, is a potent chemoattractant for smooth muscle cells and induces smooth muscle cell migration and proliferation through phosphorylation of PDGF receptor (PDGFR) and subsequent activation of the signal transduction cascade including PI3-K and p38 MAPK (mitogen-activated protein kinase) pathways (Heldin et al. 1998). Red wine polyphenols have been shown to inhibit PDGF-BB binding to βPDGFR, PDGF-BB-induced PI3-K and p38 MAPK activation, and subsequent migration of vascular smooth muscle cells (Rosenkranz et al. 2002; Iijima et al. 2002). Moreover, resveratrol binds to cellular DNA, forms a complex with Cu(II), and reduces it to Cu(I), causing redox oxygen species and producing ROS to cleave DNA strands and inhibit proliferation of vascular smooth muscle cells (Ahmad et al. 2000).
7.9 Polymorphisms of Alcohol-Metabolizing Enzymes and Cardiovascular Risk Factors
Ethanol is absorbed by the small intestine and later metabolized mainly in the liver. Alcohol dehydrogenases (ADHs) are cytosolic, dimeric, zinc-containing NAD-dependent enzymes that oxidize ethanol into acetaldehyde. When alcohol consumption is high, cytochrome P450 2E1 (CYP2E1, a member of the cytochrome P450 superfamily) can also catalyze ethanol into acetaldehyde while producing reactive oxygen species (ROS). Subsequently, acetaldehyde is converted into acetate by aldehyde dehydrogenases (ALDHs). Although there are multiple forms of ALDH in the liver, the enzyme encoded by mitochondrial ALDH2 on chromosome 12 has a very low Michaelis constant for acetaldehyde (about 1 μmol/l) and is thought to oxidize most of the acetaldehyde generated during alcohol metabolism.
Functional relevant polymorphism is known in the genes encoding ADH1 (ADH1B, Arg48His; ADH1C, Arg272Gln and Ile350Val) and ALDH2 (Glu504Lys). Exchange of amino acid in each polymorphism introduced here is based on the information obtained from the Single Nucleotide Polymorphism Database (dbSNP, http://www.ncbi.nlm.nih.gov/snp/) (build 137). The ADH1B 48Arg and homozygous ADH1C 350Val have been shown to be associated with slow metabolism of ethanol (Osier et al. 1999). ALDH2 504Lys is an inactive variant that is common among Asians but nearby absent among white populations and protects against alcoholism. Homozygous 504Lys persons are unable to metabolize acetaldehyde and drink considerably less alcohol than do homozygous 504Glu persons due to Oriental flushing syndrome characterized by facial flushing, palpitation, nausea, and headache (Brooks et al. 2009). Persons with heterozygous ALDH2 drink intermediate amounts of alcohol. The ALDH1B1 gene also shows polymorphism at two different residues of Ala86Val and Arg107Leu, though functional relevance of this polymorphism is unknown (Sherman et al. 1993). Polymorphisms of ADH1 and ALDH2 are related to outcomes of various diseases including cancer both in Asians and white populations (Seitz and Meier 2007).
Since amount of alcohol consumption is strongly related to ALDH2 polymorphism in Asians and habitual alcohol drinking is an important risk factor for hypertension, it is understandable that blood pressure is associated with ALDH2 polymorphism in Asians. According to a recent meta-analysis study including ten studies using Asian subjects, overall odds ratios for hypertension in the wild-type homozygotes and the heterozygotes vs. the null variant have been reported to be 2.42 and 1.72, respectively, and systolic blood pressure levels were 7.44 and 4.24 mmHg greater in the wild-type homozygotes and in the heterozygotes, respectively, than in the null variant (Chen et al. 2008). ADH1B variant and ALDH1B1 variant (Ala86Val) were reportedly associated with systolic blood pressure in Japanese (Hashimoto et al. 2002) and with diastolic blood pressure in Danish (Husemoen et al. 2008), respectively, though there have been only a limited number of studies on the relationships between these polymorphisms and blood pressure. In addition, a fast-metabolizing variant of ADH1B has also been shown to be associated with high triglycerides and high uric acid (Hashimoto et al. 2002). On the other hand, it has been shown that there were no significant relationships of the ADH1B variant with LDL and HDL cholesterols (Hashimoto et al. 2002; Whitfield et al. 2003). Amount of alcohol consumption was not significantly different in the three ADH1B variant groups (Hashimoto et al. 2002), and the above ADH1B-related increases in prevalence of cardiovascular risk factors might be caused by high velocity of alcohol oxidation, resulting in accelerated production of ROS. ADH1C (rs1693482) fast-metabolizing variant has been shown to be associated with an increased risk of impaired glucose tolerance/diabetes, though no clear dose-response relationship between the risk and ADH1C polymorphism was observed (Husemoen et al. 2010).
As aforementioned, flushing is a typical symptom of high sensitivity to alcohol drinking often found in Asians due to accumulation of acetaldehyde resulting from low activity of ALDH. Persons with high sensitivity to alcohol evaluated by the symptom of flushing showed a stronger association between alcohol intake and increased risk for hypertension in Japanese (Itoh et al. 1997; Wakabayashi 2005) and Chinese (Zhang et al. 2009). Thus, there is an interaction between alcohol and flushing with respect to blood pressure, suggesting that ALDH2 variant (rs671) is a risk factor for hypertension in drinkers. Similarly, HDL cholesterol and insulin resistance have been shown to be more prone to be affected by alcohol in flushers than in non-flushers by Japanese and Korean groups (Wakabayashi and Masuda 2006; Jung et al. 2010). However, a recent study has shown the opposite finding that systolic and diastolic blood pressure in Japanese heavy drinkers was higher in drinkers with the active variant of ALDH2 rs671 than in drinkers with its inactive variant (Tsuchihashi-Makaya et al. 2009). This discrepancy might be due to differences in the amount of alcohol consumption between subjects with active and inactive variants of ALDH2 rs671 in each drinker subgroup. Moderate drinkers with the homozygote of slow-oxidizing ADH1C allele reportedly had higher HDL cholesterol levels and a substantially decreased risk of myocardial infarction, suggesting a significant interaction regarding HDL cholesterol and myocardial infarction between alcohol and ADH1C (Hines et al. 2001). Interaction between alcohol and ADH1C (rs698) has also been shown regarding adiposity indices such as body mass index and waist circumference in men (Latella et al. 2009). Significant interactions have been reported between alcohol and ADH1B (rs1229984) with respect to LDL cholesterol and between alcohol and ALDH2 (rs886205) with respect to glucose tolerance (Husemoen et al. 2010).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


