Chapter 28 Airways disease
 Whatever the cause, airway narrowing leads to expiratory flow limitation, gas trapping, and hyperinflation of the lung, which manifests itself as breathlessness.
Whatever the cause, airway narrowing leads to expiratory flow limitation, gas trapping, and hyperinflation of the lung, which manifests itself as breathlessness.


Asthma
Lung diseases resulting from air pollution and infection have decreased dramatically in recent decades, but have been almost entirely replaced by asthma. It is estimated that 300 million people have asthma worldwide, with a prevalence that has increased by approximately 50% per decade. In developed countries the increasing prevalence is believed to have now levelled off at 10–15% of the population, but asthma may be continuing to become more common in the developing world (see below).1–3 In contrast to many respiratory diseases, the onset of asthma is usually in early childhood or in young adulthood. The prevalence of asthma amongst children in the developed world increased 2–3-fold in the last 50 years, though this has also now stopped increasing.4 Although the prevalence is no longer increasing, hospital admissions for asthma worldwide continue to rise, though fortunately deaths attributable to asthma have been falling consistently since the 1980s.1,5
Clinical Features
Bronchoconstriction occurs early in an asthma ‘attack’. This is particularly prominent in allergic asthma when, within minutes of exposure to an allergen, wheezing develops. Narrowing of small airways occurs due to contraction of airway smooth muscle in response to the cellular mechanisms described below. Bronchoconstriction in different regions of lung is not uniform, and positron emission tomography (PET) studies have shown that ventilation becomes patchy with clusters of poorly ventilated lung regions.6,7 This might be expected to cause maldistribution of ventilation and perfusion, but another study using PET showed that blood flow to the poorly ventilated areas is also reduced,8 presumably illustrating the efficiency of hypoxic pulmonary vasoconstriction (page 108). Heterogeneity of bronchoconstriction has significant implications for inhaled therapy, as these studies infer that most of an inhaled drug will be deposited in the better ventilated regions rather than where it is most needed.6
With more severe bronchoconstriction airway closure begins to occur during expiration, gas trapping occurs, and the lungs become hyperinflated.9 Eventually, the patient is attempting to breath in when the lungs are almost at total lung capacity, and a sensation of inspiratory dyspnoea results, even though the defect is with expiration. Physiological effects of hyperinflation are described on page 411.
Airway hyper-responsiveness (AHR) describes the observation that asthmatic subjects become wheezy in response to a whole range of stimuli that have little effect on normal individuals. Stimuli include such things as cold air, exercise, pollution (page 322) or inhaled drugs and occur via the neural pathways present in normal lungs (page 50). Methacholine or histamine can be used to measure AHR accurately by determining the inhaled concentration that gives rise to a 20% reduction in forced expiratory volume in one second (FEV1).10 Inhaled adenosine also causes airway narrowing, but unlike histamine and methacholine it does not act directly on bronchial smooth muscle.11 Bronchoconstriction in response to adenosine involves release of mediators from inflammatory cells, so the response is sensitive to the inflammatory state of the airway. For this reason, it is hoped that adenosine provocation may prove useful for monitoring the effectiveness of anti-inflammatory treatment of asthma patients, or even for differentiating between asthma and COPD.11
Cellular Mechanisms of Asthma12–14
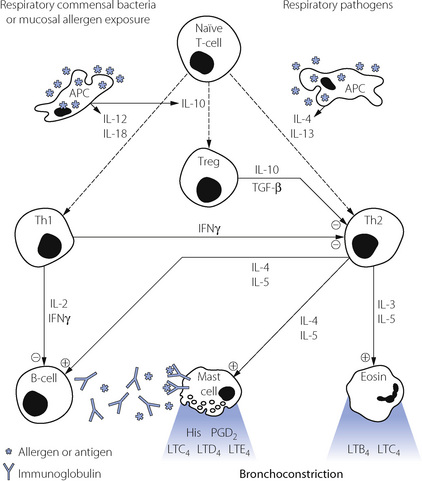
Many cell types are involved in the pathophysiology of asthma. A summary of the interactions between these cells is shown in Figure 28.1, which also shows the principal cytokines that facilitate communication between the cells.
The triggering mechanism of the mast cell is thus extremely sensitive, and is mediated by an increase in inositol triphosphate and intracellular calcium ions. Within 30 seconds of activation, there is degranulation with discharge of a range of preformed mediators listed in Table 28.1. Histamine acts directly on H1 receptors in the bronchial smooth muscle fibres to cause contraction, on other H1 receptors to increase vascular permeability, and on H2 receptors to increase mucus secretion. The granules also contain proteases, mainly tryptase, which can detach epithelial cells from the basement membrane resulting in desquamation and possibly activating neuronal reflexes causing further bronchospasm.
Table 28.1 Mediators released from mast cells when activated by IgE
| PREFORMED MEDIATORS | NEWLY GENERATED MEDIATORS | CYTOKINES |
|---|---|---|
| Histamine Heparin Serotonin Lysosomal enzymes: Tryptase Chymase β-Galactosidase β-Glucuronidase Hexosaminidase | Prostaglandin D2 Thromboxane A2 Leukotrienes C4, D4 and E4 | Interleukins 3, 4, 5, 6 and 13 Granulocyte/macrophage-colony stimulating factor Tumour necrosis factor Platelet activating factor |
The second major event after mast cell activation is the initiation of synthesis of arachidonic acid derivatives (see Figure 12.3). The most important derivative of the cyclo-oxygenase pathway is prostaglandin PGD2, which is a bronchoconstrictor, although its clinical significance is still not clear. The lipoxygenase pathway results in the formation of leukotriene (LT) C4, from which two further peptide leukotrienes, LTD4 and LTE4, are formed (see Figure 4.9).
Lymphocytes have an important role in the control of mast cell and eosinophil activation.14 Activated B-lymphocytes are responsible for production of the antigen specific IgE needed to cause mast cell degranulation. B-cells are in turn controlled by two subsets of T ‘Helper’ lymphocytes, known as Th1 and Th2 cells.
The relative activity of the opposing effects of Th1 and Th2 lymphocytes was, until recently, believed to play an important role in the development and severity of asthma. However, this convenient explanation, based mainly on studies in animals, is now thought to be an over-simplification of the situation in humans, particularly with respect to the generation of Th1 cells.15
A third sub-type of T-lymphocyte is now thought to be involved in immune regulation of the lung.14,15,16 Regulatory T-cells (Treg) are again generated from naïve T-cells, this time in response to IL-10 released by activated APCs. Activation of the APCs to produce the anti-inflammatory cytokines IL-10, IL-12 and IL-18 is believed to occur in response to antigens from respiratory tract commensal bacteria or from exposure to high levels of allergens.14 Treg cells exert an anti-inflammatory effect by secretion of IL-10 and transforming growth factor β (TGF-β), which modify the activities of both Th1 and Th2 cells.
Nitric oxide (NO) is detectable in small concentrations in the expired air of normal subjects.17 It is produced from the mucosa of the whole respiratory tract, including the nose and nasal sinuses. Nitric oxide acts as the neurotransmitter for the non-cholinergic parasympathetic bronchodilator pathway in normal lungs (page 51), is involved in control of vascular tone in all tissues and is present in blood. In asthmatic patients with active disease, NO concentration in expired air is two to ten times greater than non-asthmatics (page 414).18 In this situation, the extra NO is derived from inducible NO-synthase (iNOS, page 107) in the airway mucosa. Cytokines produced by the inflammatory cells already described are believed to result in increased production of iNOS.19
Causes of Airway Obstruction in Asthma20
Airway smooth muscle (ASM). Stimulation of bronchial smooth muscle by the substances shown in Figure 28.1 and Table 28.1 explains some of the airway narrowing seen in asthma, particularly during the acute and early stages. During deliberately induced bronchoconstriction ASM cells in asthmatic subjects also respond differently to stretching (by taking a deep inspiration) compared with in non-asthmatic subjects. In the normal lung deep inspiration causes ASM relaxation which ameliorates the bronchoconstriction, whilst in asthmatic subjects the ASM fails to respond or even contracts, exacerbating the bronchoconstriction.21 This is a poorly understood aspect of ASM physiology.22
Inflammation. Airway narrowing during the late phase response, or in severe asthma, results from inflammation of the airway. Many cytokines released during asthma have effects on blood vessel permeability and therefore cause oedema of the epithelium and basement membrane.23 Protease enzymes break down normal epithelial architecture generating defects in the epithelial barrier, leading to further inflammation and eventually detachment of the epithelium from the basement membrane. Finally, hypersecretion of mucous and impaired mucociliary clearance are both recognised features of asthma, and this correlates with the flow limitation seen in individual patients. These changes in the thickness of the airway lining translate into a significant reduction in airway cross-sectional area, and thus a large increase in resistance (page 44). Mucous, inflammatory cells and epithelial debris cause obstruction of small airways, compounded by flow limitation preventing an effective cough.
Airway remodelling.13,24,25 Repeated activation of inflammatory pathways inevitably leads to attempts by the body to repair the tissue concerned. In the lung, this results in morphological changes to both the airway smooth muscle and the respiratory epithelium. Hyperplasia of smooth muscle cells causes thickening of the airway wall even when the muscle is relaxed, and exacerbates the airway narrowing that occurs with muscle contraction because a lesser degree of muscle shortening now causes a greater reduction in the airway lumen. Goblet cell hyperplasia occurs, worsening the hypersecretion of mucous seen with airway inflammation. Finally, in asthmatic patients, there is thickening of the lamina reticularis of the epithelial basement membrane and changes to the extracellular matrix, thought to be mediated by Th2 secreted cytokines, and ultimately resulting in collagen deposition and long term loss of lung function. The clinical significance of airway remodelling in asthma is unknown, but remodelling is believed to be responsible for the long-term decline in lung function seen in some asthma patients. Airway remodelling may begin before asthma becomes severe, or is even diagnosed at all,26 and though reducing airway inflammation with steroids may delay remodelling, drugs to reverse the structural changes are as yet undiscovered.27
Aetiology of Asthma2
Genetics.28 Asthma, along with other allergic diseases, has a substantial genetic component with several genomic regions known to be linked with developing the disease.29 Environmental factors invariably contribute to the development of clinical disease, but genetic susceptibility to asthma is strong. Two reasons explain this observation. First, the genes for most of the cytokines involved in asthma are found close together on chromosome 5, and asthmatic patients may have increased expression of these, so encouraging formation of an allergic phenotype.12 Secondly, human lymphocyte antigens (HLA), which are involved in sensitisation of APCs to specific antigens, are part of the major histocompatibility complex allowing immunological ‘self recognition’, and so are inherited. It is possible that some HLA types are particularly active in the processing of common allergens and thus the stimulation of Th2 cells or the suppression of Treg cells.
A genome-wide scan of patients with asthma identified a specific gene that is strongly associated with bronchial hyperresponsiveness.29,30 The gene codes for a protein named ADAM33, part of a large family of proteins with diverse functions, including the control of cell–cell and cell–matrix interactions. In lung tissue ADAM33 protein is found in smooth muscle and fibroblasts, but not epithelial cells, indicating its possible role in airway remodelling in asthma.
Maternal allergic disease is more likely to be passed to offspring than paternal disease, though this may relate to modification of the fetal immune system in utero rather than a true genetic influence. During pregnancy, lymphocyte subsets Th1 and Th2 are closely involved in the prevention of maternal rejection, and abnormalities at this stage may influence the activity of Th1 and Th2 cells in the offspring’s immune system, leading to allergic diseases, including asthma, in later life.31
Allergy. Changes in living conditions are believed to have contributed to the increase in asthma prevalence. In the developing world, population shifts from rural to urban environments have reduced exposure to parasitic infections and increased exposure to other allergens, and it seems likely that the extensive IgE and mast cell systems that formally inactivated parasites now respond to urban allergens. In the developed world, changes in living conditions have resulted in a dramatic increase in allergen exposure, in particular house dust mite (HDM, Dermatophagoides pteronyssinus), domestic animals and fungi. Asthma is more common in affluent families, and correlates with exposure to HDM, which thrives in warm, humid houses with extensive carpeting and bedding. These conditions are ideal for the HDM and its food supply of shed skin flakes. Simply inhaling allergens is only part of the explanation of how allergen exposure causes asthma, and once again pregnancy plays a role. Allergen taken in by the mother is believed to cross the placenta and influence immunological development before birth. Neonatal T-lymphocytes taken from children who subsequently develop asthma already show a reduced production of interferon-γ in response to allergen, indicating an existing immunological susceptibility to asthma.32
Infection.33 Viral respiratory tract infections cause wheezing in many asthmatics and account for over half of acute exacerbations of asthma. In infants, respiratory syncytial or parainfluenza viruses are common, whilst in adults a ‘common cold’ rhinovirus is the most usual pathogen. Viral infection gives rise to an immune response involving many cells and cytokines, but T-lymphocytes are particularly important and undergo both virus-specific and generalised activation. Inevitably, Th2 activity is increased giving rise to wheeze and airway inflammation by the mechanisms described above (Figure 28.1). In addition, stimulation of allergic mechanisms in susceptible individuals continues for some time after the viral symptoms have subsided. Thus, for example, after a simple rhinovirus infection allergen-induced histamine production and eosinophil-induced late-phase reactions remain increased for 4–6 weeks.34
Hygiene hypothesis.35 This hypothesis to explain the rising incidence of asthma claims that in the clean, hygienic, developed world children are exposed to fewer infections or other environmental antigens than only a few decades ago. It is known that some infections may have a protective role in preventing the initiation of asthma in early childhood.33 Children who are exposed to more infections in early life, such as those with older siblings or children living on farms, are less likely to develop allergic disease. This led to a suggestion that lower infection rates in the population at large and effective immunisation programmes may have contributed to the rising incidence of asthma. Measles virus, Mycobacterium tuberculosis, respiratory and gastro-intestinal commensal bacteria, some respiratory viral infections and hepatitis A virus all have the potential to reduce asthma development by modification of the lymphocyte sub-types shown in Figure 28.1. Other micro-organisms to which the modern human is now less commonly exposed, termed ‘old friends’ by the authors,36 include lactobacilli from untreated dairy products, saprophytic mycobacteria found in mud, and helminths (worms). All three are known to promote activity of Treg cells and so potentially protect against the development of asthma (Figure 28.1). For many of these micro-organisms exposure to the entire microbe is not required and beneficial immune responses may be gained from exposure to antigens found in the dust and dirt of the environment.
Pollution. Trends in air pollution have not generally followed trends in asthma prevalence over recent decades, the levels of many pollutants declining whilst asthma becomes more common. Laboratory evidence described on page 322 describes how, in comparison with normal subjects, asthmatics develop wheeze when exposed to lower inhaled concentrations of nitrogen dioxide and sulphur dioxide. The levels required to cause wheezing are still higher than commonly encountered in the atmosphere, and though there is some evidence linking air pollution episodes to respiratory problems the effect is believed to be small.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


