Target
Action/result
Complex I
↓ Oxidative phosphorylation → ↓ ATP production
↑ ROS
Complex III
↓ Oxidative phosphorylation → ↓ ATP production
↑ ROS
Complex IV
↓ Oxidative phosphorylation → ↓ ATP production
↑ ROS
Complex V (ATP synthase)
↓ ATP production
↑ ROS
Membrane uncoupling
↓ Membrane potential (ΔΨ) → ↓ ATP production
mtDNA
Defective protein synthesis → ↓ oxidative phosphorylation → mitochondrial dysfunction
β-Oxidation
↑ Oxidative stress
Permeability transition pore
↑ Mitochondrial swelling → cytochrome C release → cell death (necrosis/apoptosis)
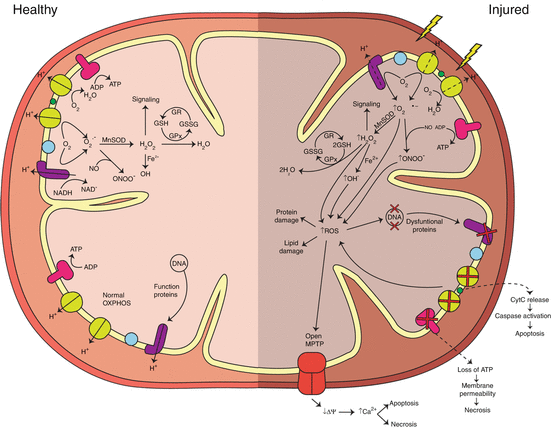
Fig. 9.1
Diagram showing the relationship between ROS generation, ATP production, and cell death pathways in a healthy mitochondrion (left side) and an injured mitochondrion (right side).
Since mitochondria are constantly exposed to low levels of ROS, they have adapted by developing an antioxidant response system to reduce the oxidant burden in the mitochondria and preserve mitochondrial integrity and function. Mitochondria are equipped with high concentrations of glutathione (GSH), superoxide dismutase, glutathione peroxidase, and other antioxidant enzymes to remove potentially harmful oxidants; however, even in mature mitochondria, increased production of ROS can quickly overwhelm these systems [14]. The expression of many of these systems changes over the course of postnatal lung development, potentially endangering children vulnerable to increased damage due to pulmonary damage during this period (Table 9.2). The expression of those systems are again changed with advancing age due to cumulative lifetime protein and DNA damage that can alter activity or decrease protein expression, which can result from the failure of multiple systems. Animal studies have shown that supplementation with ascorbic acid and other antioxidants are capable of protecting the lung against age-related oxidative mitochondrial DNA (mtDNA) damage [9].
Table 9.2
Age-related factors that alter susceptibility to pulmonary cellular injury
Immature | Aged |
|---|---|
Incomplete development of detoxification enzyme systems | Damage to enzyme systems |
Increased cellular proliferation | Reduced repair/regeneration capacity |
Increased breathing frequency | Cumulative oxidative damage to DNA, proteins, and lipids |
Increased lung surface area/body weight | Decreased immune function |
Immune system is not fully developed | Reduced mitochondria turnover |
Incomplete airway and alveolar development | Reduced airway clearance mechanisms |
Morphological changes in mitochondrial structure | Increased caspase activity |
During postnatal lung development, mitochondrial volume increases and morphological alterations occur primarily in nonciliated epithelial cells and type II alveolar cells, two proliferating cell populations in the airways and alveoli [15, 16]. In the rabbit and rat, mitochondrial volume increase during both pre- and postnatal development in the nonciliated bronchiolar cells, including Clara cells [15, 17]. In prenatal rabbits, less than 15 % of the volume in Clara cells is mitochondria; however, by 4 weeks postnatal, Clara cells resemble that of older animals, indicating that mitochondria undergo a shift in abundance during differentiation [18]. In the rat, during postnatal development, mitochondria change from a single spherical organelle to a complex branched structure in type II alveolar cells [16]. Changes in mitochondrial size and intracellular distribution during cellular differentiation may alter cellular responses to injury and alter the ability of the cell to recover from toxic insults. In contrast to the changes that occur during development, advanced age also alters mitochondrial dynamics. The mitochondrial theory of advanced aging has been widely accepted since its introduction in the 1970s and is believed to be the driving force behind cellular aging in the body [19, 20]. Age-related changes in basic mitochondrial functions have been reported, including decreased respiratory control ratios (RCR), changes in mitochondrial membrane permeability (ΔΨ), and activities of the electron transport chain complexes (I, III, and IV), which are believed to be the result of cumulative oxidative damage [21].
Mitochondrial DNA (mtDNA)
Mitochondrial DNA (mtDNA) is a double-stranded circular DNA that encodes the mitochondrial genome and resides in the mitochondrial matrix near the electron transport chain. In humans, the mtDNA genome has 16,569 bp and contains 13 protein-coding genes, most of which are electron transport chain (ETC) proteins. Other animals, including mice, rats, and primates, have slight variations in the size of the DNA (16.3–16.5 kb) [22]. mtDNA lack introns, so mutations in mtDNA are likely to affect the coding regions which may result in the production of dysfunctional proteins [23]. In contrast to nuclear DNA, mtDNA lack protective histones and therefore are at increased risk for oxidative damage from the ETC, the primary source of ROS in the mitochondria [24]. With this proximity to the oxidant producing ETC, mtDNA is prone to oxidative damage that can alter protein synthesis of critical proteins in the ETC and ultimately decrease the ability of cell to produce energy. To protect the mtDNA, a combination of DNA repair mechanisms and degradation of irreparable mtDNA are used to maintain integrity [25]. Unlike damaged nuclear DNA, replication of damaged mtDNA is not blocked, allowing defective or nonfunctional proteins to be synthesized and inserted into the ETC [26]. Oxidative lesions of mtDNA also accumulate with age in rodents [27] and humans [28], with levels of mtDNA damage being significantly greater than those found in nuclear DNA [9]. Under normal physiologic conditions, it is estimated that DNA damage ranges from 1 modification for every 130,000 bases of nuclear DNA to 1 in 8,000 bases for mtDNA [29]. mtDNA damage is also correlated with oxidant-mediated cell death [30, 31]. mtDNA deletions in conjunction with membrane lipid peroxidation may also play a key role in mitochondrial dysfunction due to aging [27].
In the mouse lung, mtDNA content increases during the first 2 months of postnatal development, drops slightly around 5 months, and increases again by 15 months of age, showing that the cyclic increase of mtDNA levels closely correlates with two periods of increased susceptibility to oxidative stress and injury, during early life and advanced age [32]. In humans, mtDNA content increases by 2.6-fold in lung tissue from patients over 80 as compared to those less than 20 [28]. Tissues from aging and senescent rats have been shown to contain increased mtDNA as compared to younger age groups [27]. mtDNA lesions increase in postmitotic tissue as compared to mitotic tissues in humans; however, one study using Fisher 344 rats reveals the opposite effect [33].
Mitochondrial Dysfunction
Mitochondrial dysfunction occurs when the mitochondrial function is impaired resulting in reduced energy production. When electron transfer between oxidoreductase complexes becomes impaired, the matrix becomes a more electron-rich environment, which increases autoxidation, increases ROS generation, and decreases respiration [34, 35]. Provided that energy production is essential for cell survival, any dysfunction that occurs in the mitochondria has the potential to affect cellular homeostasis and shift toward cell death.
In the mitochondria, two common measures of respiratory function are state 3 and state 4 respiration [36]. State 3 respiration refers to the state of actively respiring mitochondria in which ADP is converted to ATP. In contrast, state 4 respiration is the slower rate of respiration that occurs after ADP has been depleted. Mitochondria are believed to function between state 3 and state 4 in vivo. The RCR is a ratio of state 3 to state 4 respiration which serves as an index of tightness of coupling between respiration and phosphorylation. The relationship between ATP synthesis (during the respiratory burst of state 3 respiration) and oxygen consumption is determined by the P:O ratio [37].
In the lung mitochondria of senescence-accelerated mice, advanced age results in reduced activity of the electron transport chain complexes (I–IV) and ATP production [38]. In Fisher 344 rats, using succinate as a substrate, state 3 respiration, uncoupled state respiration, and respiratory control ratio (RCR) are all significantly decreased in aged rats as compared to adults [39]. Following exposure to 3 ppm ozone for 8 h, respiration rates for state 3, state 4, and the uncoupled state, as well as the RCR and ADP/O ratios, decrease similarly in both adult and aged rats [39]. This same study also compares isolated heart and lung mitochondria and shows a greater decrease in metabolism in lung mitochondria. This may be attributed to proximity of ozone-derived products and their ability to interact with lung mitochondria. Ozone also induces changes in the presence of NAD- and FAD-linked substrates that may indicate alterations in the inner mitochondrial membrane. Mitochondria may be a subcellular organelle target for ozone interactions and that alteration of energy metabolism may contribute significantly to the overall damage caused by this oxidant. Impaired energy production in the aged mitochondria may contribute to the age-related physiological consequences and accelerated alterations during chemical stress [39]. Compared to 5-month-old mice, 10-month-old mice show decreased activity in complexes 1, 2, and 4 in both male and female mice, while complex 3 is depressed only in male mice [38]. Additionally, ATP production and ATP/ADP ratios are depressed in the 10-month-old mice, indicating that mitochondria in the aged mice are not as efficient in production of energy as the adult mice [38]. State 3 and state 4 O2 consumption is significantly higher in immature rats compared to both adult and aged rats; however, O2 consumption is not significantly different between adult and aged groups. RCR remains constant across all age groups. H2O2/O2 consumption in state 4 is significantly lower in the young and aged rats as compared to the same in adults [40].
General Considerations for Lung Development and Aging
The adult lung is comprised of many morphologically distinct cell phenotypes organized into a highly branched series of tissues surrounding the air passages. Each developmental stage of the lung is associated with the transformation from a simple tubular structure into a highly complex organ system that is potentially susceptible to modification by toxic agents. As a result, several aspects of the developmental process should be considered when evaluating the potential toxicity of a compound. First, it is important to remember that lung development is a multi-event process which is not restricted to prenatal life. Although the development of the tracheobronchial bud begins early in gestation, a large amount of lung growth and therefore cellular differentiation occurs during the postnatal period. Second, only a restricted number of maturational events must be complete at birth for successful postpartum survival of an organism. Third, there are three general categories of events that occur throughout the pre- and postnatal development periods – branching morphogenesis, overall growth, and cellular differentiation – that may alter the toxicity of a compound in the lung. The process of branching morphogenesis begins with the formation of the tracheobronchial bud. This branching continues until there is an extensive airway tree of about 22 generations in humans. The formation of vascular trees, arterial and venous, and capillary beds associated with both the conducting airways and the gas-exchange region occurs at the same time as the airway tree. The highly complex structure of the gas-exchange area also requires an extensive period of branching morphogenesis to form alveolar saccules and ducts, most of which occurs during the postnatal period. Formation of the interalveolar septa, a process termed “alveolarization,” begins during late gestation and continues through the first 2 years of life in humans [41, 42]. During the alveolarization period in rats, type II cells undergo a series of morphological changes, including a shift from single spherical mitochondria to more complex branched structures [16]. The overall size, or volume, of the lungs and trachea increases in a steady progression throughout pre- and postnatal life until body size ceases to increase. The cells of the airway tree, matrix, and interstitium differentiate at varying rates. Fourth, all of these developmental events occur in combination with a steady increase in total cell mass, primarily through cellular proliferation.
Even though federal agencies have identified children as a susceptible population, the impact of toxicants during developmental of the lower respiratory tract is not well understood. One reason is that the mechanisms regulating the complex series of morphogenetic and events of differentiation are only now beginning to be identified. Some of the regulatory factors, such as hormones and cytokines, have been recognized, but epithelial-matrix interactions and cell-cell (epithelial-epithelial or epithelial-mesenchymal) interactions are only starting to become evident. Toxicants can modify developmental events in many ways. For example, exposure to a toxicant can result in a poorly functioning, or inadequate, gas-exchange system. Toxicants can also induce a partially differentiated cell population to develop phenotypes not found in uninjured lung or alter the expression of phenotypes present so they are more or less resistant to injury.
Once development and growth is finished, we must then consider the issues associated with the aging of the lung. With increasing age, numerous physiological changes occur in the lung, including reduced ventilatory control, respiratory mechanics, and gas exchange [43]. In the aged lung, respiratory muscle strength decreases resulting in impaired ability to cough, which is an important mechanism for airway clearance [44]. Additionally, aging alters deposition, retention, and clearance of particles [44]. Alterations are noted in structural components of the airway and alveolar wall, indicating impaired repair mechanisms, many of which are associated with age-related lung diseases [44, 45]. One of the driving forces behind aging is believed to be cellular senescence. Senescence is a state in which cellular proliferation is arrested and cells also become resistant to apoptosis [46]. Typical responses to DNA damage enable the cell to repair the damage when possible; however, if the cell is unable to do so, it will respond by arresting cell-cycle progression [47]. Senescent cells have been shown to increase expression of genes, including extracellular matrix degrading enzymes, inflammatory cytokines, and growth factors [46]. These factors disrupt normal tissue structure and function and also affect non-senescent cells by causing abnormal responses to stimuli [46]. In rat lung and liver, advanced age is associated with increased activation of executioner caspase-3, caspase-6, and caspase-7 and progression of apoptosis [48]. Additionally, advanced age also increases activity of caspase-2 and caspase-9, both of which are involved in mitochondria-mediated apoptosis in liver suggesting that as cells age, mitochondria-centric cell death pathways may be more active.
Cellular Antioxidant and Detoxification Systems
The mature lung has well-developed antioxidant and xenobiotic metabolism systems to detoxify endogenous and exogenous compounds that have the potential to cause cellular injury. Many of these detoxifying enzyme systems are confined to subcellular compartments (i.e., cytoplasm, mitochondria, endoplasmic reticulum); however, properly functioning systems in all compartments are essential for protecting the cell as a whole. The following sections describe the development of these systems during early life and the loss of activity with advanced aging that may alter susceptibility to cellular injury in these age groups.
Age-Specific Expression of Antioxidant Enzyme Systems
An obvious role of antioxidants in the lung is protecting the lung from oxidative stress. This level of protection is critical at birth, when the lung switches from a relatively hypoxic state to a relatively hyperoxic state. Additionally, as the lung ages, oxidant burden continues to increase. The antioxidant enzymes [superoxide dismutase (SOD), catalase, and glutathione peroxidase (GPx)] and nonenzymatic antioxidants (glutathione, GSH) also have an important role in modulating cellular interaction with environmental toxicants such as ozone and nitrogen dioxide. During late gestation, changes in the fetal lung include the development of the antioxidant enzymes, including SOD, catalase, and GPx. These enzymes play an important role in the detoxification of highly reactive oxygen species that are produced during normal aerobic cellular respiration as well as during oxidant injury. In general, the pulmonary antioxidant enzyme system develops during the last 10–15 % of gestation in humans [49] as well as in many laboratory animal models including rats, hamsters, guinea pigs, rabbits, and lambs [50–58].
Superoxide Dismutase (SOD)
The antioxidant enzyme SOD rapidly catalyzes the conversion of the superoxide anion to hydrogen peroxide and oxygen. There are three isoforms of SOD: copper-zinc SOD (Cu/ZnSOD), manganese SOD (MnSOD), and extracellular SOD (EcSOD), all of which are found in the lung. MnSOD, an inducible form of superoxide dismutase, is predominantly located in the mitochondria [59, 60], while the noninducible Cu/ZnSOD is located in the cytosol [61]. EcSOD is localized in the extracellular matrix, vessel walls, and extracellular fluid of the lung [62, 63]. EcSOD is thought to play a role in modulating nitric oxide concentrations by regulating the amount of superoxide anion available to react with it [64], but very little is known about this form in the developing lung [65].
In most animal models, SOD activity increases throughout the pre- and postnatal periods; however, there are some species differences (Fig. 9.2). In rats, pulmonary Cu/ZnSOD activity and total enzyme content peak during the late gestational period (last 10–15 % of gestation) and activity peaks again early in the postnatal period, finally reaching adult levels at 4 weeks [54, 56, 67, 68, 72]. Cu/ZnSOD activity is elevated in young rats compared to adult and aged rats (40). Temporal expression of mRNA does not correlate with the activity levels [68, 73]. In contrast, pulmonary MnSOD activity and mRNA content remain steady throughout late gestation and the early postnatal period in the rat [51, 72, 73], even though total enzyme content increases 6.9-fold [54]. In the mouse, Cu/ZnSOD mRNA expression peaks at 15-day gestation, is low during the later stages of fetal lung development, and peaks again at birth [73, 74]. In rabbits, pulmonary Cu/ZnSOD and MnSOD activities also peak in late gestation and in the postnatal period [55]. In the rabbit, EcSOD expression is low and contained within the intracellular compartment during the fetal period. Expression increases and shifts to the extracellular compartment with increasing age [65]. In the developing rat, EcSOD protein expression and activity increase from 2 days of age until adulthood [75]. In contrast to experimental animals, human Cu/ZnSOD and MnSOD activity remains constant during fetal and neonatal lung development, and no surge in expression is seen [49, 66].
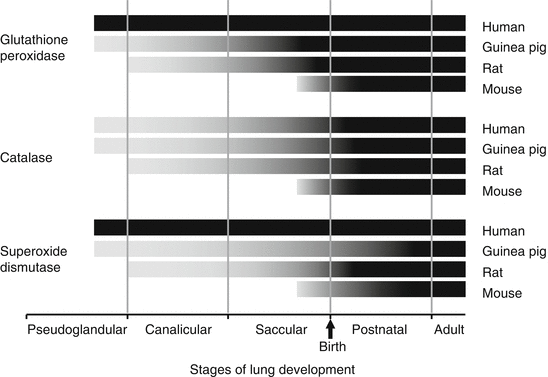
Fig. 9.2
Species-dependent expression of antioxidant enzyme protein and/or activity during stages of lung development. Gray portion of bars represent transitional expression, black portions of bars represent mature expression for human [49, 66], guinea pig [55], rat [50, 54, 67–70], and mouse [291] (Reprinted with permission from Fanucchi [71])
In the aged lung, SOD activity and protein expression results have been across the board. In one study, SOD activity (total SOD, Cu/ZnSOD, and MnSOD) is not altered in the lungs of aged rats [76]. Another study found that MnSOD and Cu/ ZnSOD activities increase in aged rats (26 months) as compared to young (4 months old) rats; however, mRNA expression is not altered by age [77]. While some of the results are mixed, majority of the literature has shown that SOD protein expression and activity decrease in the lung tissue of aged rats [78–81].
Glutathione Peroxidase (GPx)
GPx is a cytosolic enzyme that catalyzes the reduction of hydrogen peroxide to water and oxygen as well as catalyzing the reduction of peroxide radicals to alcohols and oxygen [52]. The oxidation of GSH to GSSG provides energy for this catalysis, but the higher oxidation state can be toxic, because NADPH is consumed when GSSG is reduced back to GSH in cells.
As with SOD, GPx activity and total enzyme content increase during late gestation in two of the experimental animals studied: guinea pig and rat [54, 56, 72, 73] (Fig. 9.2). However, mRNA expression steadily decreases during fetal development and does not correlate with activity levels [51, 73]. GPx activity in postnatal rats is lower than adults, suggesting that GPx activity increases during the postnatal period [40]. In the mouse, however, both GPx activity and mRNA levels peak in late gestation and then activity steadily decreases throughout the postnatal period [20, 74]. In humans, GPx activity is lowest at 10–20 weeks of gestation, is highest during the early postnatal period (41–50 weeks postconception), and drops at 3–5 months of age [49, 82].
Catalase
Catalase, like GPx, accelerates the combination of two hydrogen peroxide molecules to produce water and oxygen. Catalase activity steadily increases during both the fetal and postnatal periods of lung development in humans [49] and in all experimental animals studied, including guinea pigs, rats, and mice [20, 54, 56, 72, 73] (Fig. 9.2). In contrast to GPx, the catalase mRNA expression during this time is consistent with the increase in activity [73]. In humans, catalase activity increases 3.5-fold between 10 weeks of gestation and 3 months of postnatal age, unlike SOD and GPx, which remain constant throughout human lung development [49]. The increase in catalase activity during the late gestational period has been suggested by McElroy and colleagues to be linked to maturation of the surfactant system and has been correlated temporally to increases in lung dipalmitoylphosphatidylcholine (DPPC) content [49].
The effects of advanced aging on catalase activity are mixed. In rats, mRNA expression is significantly lower in the aged group as compared to the young rats [77]. Catalase activity has been shown to decrease in aged 129/ReJ mice [83] and rats [78, 79, 81]. However, other studies have shown catalase activity unaffected by advanced age in C57BL/6 K mice [83] or rats [40, 77]. In one study, catalase activity is found to be increased in the lungs of aged BALB/c mice [83].
Glutathione
Glutathione (γ-glutamylcysteinylglycine, GSH), a tripeptide, acts as an antioxidant by directly scavenging free radicals through the donation of a hydrogen atom. In the lung, GSH is found in both intra- and extracellular compartments. In the cell, 85–90 % of the GSH resides in the cytosol, 10–15 % localized in the mitochondria, and small percentages in the endoplasmic reticulum and nucleus [84]. The synthesis of GSH is homeostatically controlled, both inside the cell and outside [85–88]. GSH is not taken up by pulmonary cells in its intact form. It is synthesized and utilized through the γ-glutamyl cycle, a multistep, ATP-dependent process. Very simplistically, it begins with the cleavage of extracellular GSH and other glutamate-containing peptides by γ-glutamyl transpeptidase, a membrane-bound enzyme. The resulting amino acids can then be moved by specific transporters into the intracellular environment [89]. The rate-limiting step in the synthesis of glutathione, however, is the formation of γ-glutamylcysteine by γ-glutamylcysteine synthetase (γ-GCS). The formation of GSH is complete when GSH synthetase catalyzes the addition of glycine to γ-glutamylcysteine to form γ-glutamylcysteinylglycine (GSH) [85]. Even though GSH plays an important role in protection against both oxidant and reactive metabolite injury, there is very little information available on glutathione levels or γ-glutamyl cycle enzyme activity during fetal and postnatal lung development.
Evidence from the few rodent studies available suggests that neonates have similar GSH levels as adults. In rats, pulmonary GSH levels are at mature levels right before birth, decrease 30 % at birth, and then increase back to mature levels between 10 and 15 days [90, 91]. The activities of two important enzymes of the γ-glutamyl cycle, γ-GCS and glutathione synthetase, are similar in the lungs of 1–2-week-old mice and adult mice [20]. As observed in the mice, there are no differences in the γ-GCS activity among the human fetal (15–19 gestational weeks), neonatal (26–42 gestational weeks), and adult lungs [69]. γ-Glutamyl transpeptidase mRNA is detectable by polymerase chain reaction in late fetal and early postnatal rat lung and is located in the type II cell [92]. However, pulmonary γ-glutamyl transpeptidase activity is very low in newborn rat, and only gradually increases to mature levels [92, 93]. In the late postnatal and adult rat lung, γ-glutamyl transpeptidase is found in higher abundance in Clara cells than in type II cells [92]. Pulmonary GSH synthetic activity is affected by several factors. One factor, maternal nutrition, has been shown to be very important in regulating the perinatal activity of GSH cycle enzymes. Pulmonary γ-GCS activity is lower in rat pups from dams that were fed low-protein diets than in pups from dams fed diets supplemented with casein [93]. Surgical procedures in newborn guinea pigs also affect GSH synthetic activity and result in increased pulmonary GSH content [94].
The effects of aging on tissue GSH levels rats are mixed. One study demonstrates that the status of reduced glutathione (GSH) is not different between the lungs of adult and aged rats [90]. However, multiple studies have shown a decrease of GSH in the GSH pool in the lung [79–81, 95, 96]. In the mouse lung, the ratio of GSSG (oxidized disulfide form of GSH) to reduced form of glutathione (GSH), a measure of oxidative capacity of the lung tissue, is increased with age suggesting the decreased ability to manage oxidative stress [96]. In the senescence-accelerated mouse, GSH/GSSG ratios decreases in the lung mitochondria with advanced age suggesting a reduced availability of GSH in this compartment [38]. The ability to synthesize and maintain reduced GSH decreases with age. GCL has also been shown to decline in the lung with advanced age. GSH reductase, however, is not affected by age [96]. In addition to alterations in tissue levels, GSH in the epithelial lining fluid decreases with age in humans [97] and mice [96]. The mitochondrial pool of GSH has been shown to be more oxidized with age, thereby reducing its protective effect in the mitochondria [98]. This change is greater in the mitochondria than in the whole cell [9].
Miscellaneous Antioxidants
Little information is available regarding the expression of other antioxidants during lung development. Ceruloplasmin, a free radical scavenger, is a major extracellular antioxidant in human lung epithelial fluid [99]. Ceruloplasmin mRNA has been detected in fetal lung of species as varied as mouse and baboon [100]. The mRNA first appears in baboon lung during the pseudoglandular stage (60-day gestation) in bronchial epithelium. By the saccular stage of development (140-day gestation), ceruloplasmin mRNA is detected in all airway epithelium. In mice, ceruloplasmin mRNA is detected during the saccular stage and is expressed in epithelium of all airways. Peroxiredoxin, an antioxidant which reduces hydrogen peroxide to molecular oxygen has also been described in lungs of newborn baboons [101]. Both pulmonary peroxiredoxin mRNA and activity are expressed at low levels in fetal baboons and are upregulated by oxygen treatment. Urate and ascorbic acid (vitamin C) levels in bronchoalveolar lavage fluid have been described in preterm human infants [102]. Urate levels drop during the first 2 weeks of life, while ascorbate levels drop during the first week then increase during the second week of life. Ascorbic acid levels decrease again with advanced age [81, 103, 104]. Aging also reduces α-tocopherol levels in the lung [81].
Age-Specific Expression of Xenobiotic-Metabolizing Enzyme Systems
The interplay and balance between the xenobiotic-activating enzymes and xenobiotic-detoxifying enzyme systems have been shown to be critical factors in dictating the toxic response of the respiratory system to bioactivated compounds in adults [105–108]. This interplay is also important in perinatal animals during pulmonary morphogenetic and differentiation processes. Xenobiotic-metabolizing enzymes act on compounds to make them more water-soluble and increase their rate of elimination. Many lung-targeted toxicants, such as furans [109], chlorinated hydrocarbons [110, 111], aromatic hydrocarbons [108, 112], indoles [113–118], pyrrolizidine alkaloids [119], naphthalene [106, 120, 121], and butylated hydroxytoluene (BHT) [122–124], require bioactivation in order to exert their toxicity. The reactive metabolites of these bioactivated compounds are then detoxified by a number of pathways. Other compounds, such as organometallic [125] and amphiphilic agents [126], are toxic when introduced into the body and also require metabolic detoxification.
Cytochrome P450 Monooxygenases
The cytochrome P450 (CYP) monooxygenases are important enzymes in both bioactivation and detoxification of compounds. In adults, immunoreactive CYP enzymes have been detected in four pulmonary cell types: the nonciliated bronchiolar (Clara) cell, the type I and type II epithelial cells, the endothelial cell and the alveolar macrophage. The developmental expression of CYP monooxygenases is the most extensively studied of all the pulmonary xenobiotic-metabolizing enzymes. Even so, the expression of pulmonary CYP monooxygenases in the differentiating lung has been evaluated in only five species: rabbit, hamster, mouse, rat, and goat [120, 127–135]. Some of the CYP isozymes (and representative substrates) that have been evaluated during lung development include CYP1A1 (ethoxyresorufin), CYP2B (O,O,S–trimethylphosphorothioate), CYP2F (naphthalene), and CYP4B (2–aminofluorene). In addition, NADPH CYP reductase, important for electron transport during the catalytic cycle of CYP monooxygenases, has also been evaluated. In general, the first detectable expression of CYP monooxygenases occurs after the development of the smooth endoplasmic reticulum (SER). Protein for NADPH CYP reductase is detected before the monooxygenase isozymes in all species evaluated. The youngest age at which the intracellular protein can be detected immunohistochemically varies substantially within the five species evaluated (Fig. 9.3). Isozymes CYP2B and CYP2F are expressed earliest: they are detectable in the late fetal stage in mice and hamsters [129, 135], in the early postnatal period of rabbits and rats [131, 136, 137, 139], and around 6 weeks of age in goats [127, 128]. CYP4B is detected 2–3 days of age later. Activity for these proteins is first detected approximately 2–3 days after the protein is immunologically detectable. The temporal and spatial distribution of immunoreactive pulmonary CYP protein has been described in detail for the rabbit [136]. Initially, immunoreactive protein is detected only in the most apical border of a small percentage of the nonciliated cell population. During the period of time in which the amount of detectable protein increases, the distribution changes in two ways. First, an immunologically detectable protein is found in an increasing proportion of the nonciliated cells. Second, the distribution of detectable protein within an individual cell moves from the apex to the base until it is evenly distributed throughout the cell. The timeframe between when the protein is first detectable and when it reaches mature distribution and intensity also varies among species. Mature expression of CYP proteins can take up to 4 weeks in rabbits and mice [129, 136], 3 weeks in hamsters [135], and as little as 1 week in rats [132, 137]. Mature expression of the immunoreactive protein, however, does not necessarily indicate mature enzyme activity, especially in rats and rabbits [132, 137]. This suggests that the activity of these enzymes continues to increase after the protein density and organelle composition have reached adult levels. CYP3A is an important isozyme in the metabolism of many therapeutics and environmental pollutants as well as endogenous compounds that humans are exposed to daily [140] although most of the literature focuses on hepatic expression. Comparative studies of the postnatal pulmonary expression of isozyme CYP3A in the rat [141] indicate that this CYP activity is detectable in postnatal lung as well as adult lung. It is critical to understand the temporal and spatial development of CYP monooxygenase activity in order to be able to extrapolate among species.
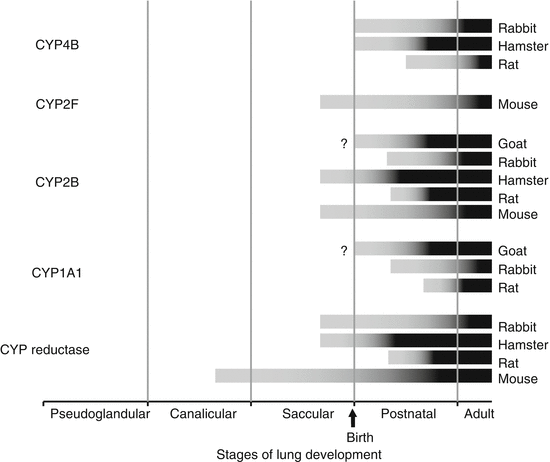
Fig. 9.3
Species-dependent expression of CYP monooxygenase protein and/or activity during stages of lung development. Gray portion of bars represent transitional expression, black bar portions of bars represent mature expression for goat [128], rabbit [136], hamster [135], rat [132, 137], and mouse [138]. Question marks (?) indicate lack of available information during the fetal period (Reprinted with permission from Fanucchi [71])
Glutathione Transferase
It is essential to understand the temporal and spatial development of the detoxifying enzymes, including glutathione transferase, in order to appreciate the role they play in protecting the lung during development. Glutathione S-transferases (GSTs) are a group of dimeric enzymes present in multiple subcellular compartments, including the cytosol, mitochondria, and other membrane-bound organelles (including the endoplasmic reticulum and nucleus) [84] that catalyze the conjugation of GSH to electrophilic compounds. In human tissues, GSTs are grouped into seven classes [142–146]. Many of the classes have been described in adult lung, although only the expression of GST α, μ, and π has been evaluated in the developing lungs of mice [147, 148], rabbits [149], goats [127], guinea pigs [149], and humans [150–153] (Fig. 9.4). Unlike CYP monooxygenases, which are expressed in only four cell types in the lung, pulmonary GSTs are expressed in multiple cell types including both ciliated and nonciliated bronchiolar epithelial cells, alveolar epithelial cells, endothelial cells, and smooth muscle cells. The ubiquitous distribution of the transferase enzymes suggests a role in protecting cells from reactive intermediates from both exogenous and endogenous metabolism. In goats, rabbits, and guinea pigs, information regarding the expression of GSTs is available for only the postnatal period of lung development [127, 148]. Rabbits and mice at birth have pulmonary GST protein expression and activity levels similar to adults, while GST expression is not mature in goats until later in the postnatal period. In mice, the only laboratory animal in which GST expression has been described during fetal lung development, GST isozymes are present very early in development; however, the isozymes do not reach mature expression levels until early in the postnatal period [148]. In contrast to mice, human fetal lung has been shown to contain more overall GST activity than adult lung [153]. During human lung development, the total activity of pulmonary GST decreases by fivefold between 13 weeks gestation and birth and then remains constant [152]. Pulmonary GST (cytosolic) is significantly lower in weanlings than adult horses, suggesting that GST system continues to develop during the postnatal period [156]. Pulmonary GST activity is due to a combination of all the isozymes, and the contribution of each isozyme changes during development. In humans [150, 157, 158], GST1 (μ) increases as a percentage of total GST activity over development, from 5 % at 10 weeks of gestation (pseudoglandular stage of lung development) to 25 % at birth and 60 % in adult tissue. GST2 (α) contributes 50 % of the total activity consistently. GST3 (π) decreases as a percentage of total activity over development, from 50 % of the total GST activity at 10 weeks of gestation to 15 % at birth, and is almost undetectable in adult tissue. These activity data are supported by immunohistochemical data [151]. GST2 is expressed consistently throughout development: strongly in proximal airway epithelium and weakly in more distal airways in fetuses, newborns, and adults. GST3 decreases in expression during development. It is expressed strongly in all epithelial cells in early gestation, and then expression is lost in the distal airways by 24 weeks of gestation [151, 153, 159].
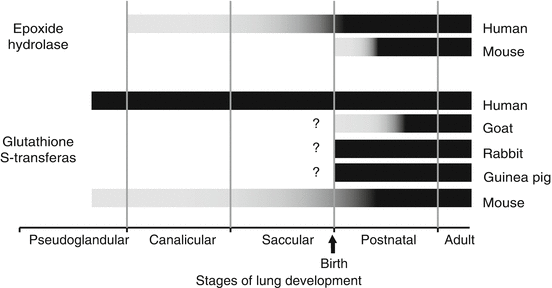
Fig. 9.4
Species-dependent expression of glutathione S-transferase (alpha, mu, and pi combined) and epoxide hydrolase (mEH and cEH combined) protein and/or activity during stages of lung development. Gray portion of bars represent transitional expression, and black portions of bars represent mature expression for goat [127], rabbit [149, 292, 293], guinea pig [149], mouse [148], and human [150–155, 294]. Question marks (?) indicate a lack of available information during the fetal period (Reprinted with permission from Fanucchi [71])
Epoxide Hydrolase
There are three general forms of epoxide hydrolase (EH) which all create 1,2-dihydrodiols from epoxides, although each form has different substrate specificities and tissue distribution [160]. The first form, microsomal EH (mEH), is involved in the conversion of cyclic epoxides and is found in high levels in the smooth endoplasmic reticulum (SER) and in lower levels in other membranous organelles. The second form, cytosolic EH (cEH), hydrates aliphatic epoxides and is found in the cytosol and peroxisomes. The third form, cholesterol EH, catalyzes the hydration of cholesterol epoxides and is located in the microsomal fraction. Substrate specificity may also differ immensely among species and tissues [160]. As with most metabolic enzymes, the highest levels of both mEH and cEH activity are found in the liver, and this is true in adults of a wide variety of species: rabbits [161], humans [161], rats [162], and mice [148, 163]. The ratio of cytosolic to microsomal EH activity, though, varies by species and by organ. In humans, cEH activity is higher than mEH activity in both the liver and the lung [154, 155, 159, 164]. In mice, while cEH activity is higher than mEH activity in the liver, the reverse is true in the lung [164]. EH activity also varies within the lung. EH activity is reported to be present in microdissected airways of dogs with the highest activity in distal airway generations and lower activities in proximal airway generations [165].
There are few studies on the development of epoxide hydrolase. In mice, the predominant pulmonary form of epoxide hydrolase is microsomal, with the cytosolic form found only in vascular smooth muscle [148]. Immunoreactive mEH protein is detectable in airway epithelial cells soon after birth and reaches mature density and distribution near the age of weaning (Fig. 9.4). However, specific activity for mEH in mouse lung is detectable at mature levels at 7-day postnatal age [148]. In contrast to the mouse, the predominant pulmonary form of epoxide hydrolase in humans is cytosolic and EH mRNA expression gradually increases to adult levels by 65 days postnatal [134]. Increases in fetal human pulmonary EH activity do not correlate with increases in mRNA expression [154]. Pulmonary soluble epoxide hydrolase did not vary with age in horses [156]. In mice, mEM activity decreases from 3 to 12 months of age followed by periods of increased and decreased activity through 30 months; however, cEH activity increases during the first 9 months of life followed by a steady decrease through 30 months of age [166].
Glucuronyl Transferase
Glucuronidation is also a major pathway for the metabolic elimination of parent compounds (such as 4-ipomeanol) or primary metabolites (such as 1-naphthol) and as such can provide important means of protecting extrahepatic tissues from toxicants [167]. As with the other phase II enzymes discussed in this chapter, glucuronyl transferases are widely distributed throughout tissues, with the highest activity found in the liver. These enzymes have been described in the lungs of adult rabbits [168], dogs [165, 169], humans [164], and sheep [170]. Similar to CYP monooxygenases, the distribution of UDP-glucuronyl transferase is restricted to the bronchial epithelial cells, Clara cells, and type II pneumocytes in the rat lung. There are reported species-specific differences in activity levels. UDP-glucuronyl-transferase activity has been shown to be evenly distributed throughout the respiratory tract of the dog and is similar to activity levels found in the liver [165]. In the human [171], pulmonary UDP-glucuronyl transferase activity is considerably lower than hepatic activity. In contrast to the detailed studies of the development of hepatic glucuronyl transferases, there is only one report on the developmental expression of this enzyme in the lung. In goats, pulmonary UDP-glucuronyl transferase activity has been reported to be lowest at birth and highest in adults. However, the pattern of enzyme expression also varies by substrate used [127].
Miscellaneous Enzymes
In addition to the four enzyme systems already mentioned, there is a small amount of information available concerning the expression of two other enzymes during lung development: flavin-containing monooxygenases and sulfotransferases [139, 172]. Flavin-containing monooxygenases are important oxidative metabolizing enzymes and have much in common with the cytochrome P450 monooxygenases. They have similar molecular weights, are localized in SER, have the highest expression in liver, require NADPH, and have multiple isozymes [173, 174]. To date, flavin-containing monooxygenases have only been studied during lung development in rabbits [139]. Activity, protein, and mRNA are all expressed as early as 25-day gestation (canalicular stage). Flavin-containing monooxygenase expression is high prenatally (except for an unexplained decrease at 28-day gestation), drops immediately after birth, and then steadily increases throughout postnatal lung development. This expression pattern matches that of CYP2B4 and CYP4B1 in the rabbit lung [139]. Sulfation is a major detoxification pathway, resulting in a highly water-soluble product [175, 176]. There are two major subfamilies: phenol sulfotransferase and hydroxysteroid sulfotransferase, each of which has different substrate specificities [177]. Human pulmonary hydroxysteroid sulfotransferase, which sulfates steroids and cholesterol, is found in low levels early in gestation (around 56-day gestation) and expression peaks at 1 year after birth [172]. Hydroxysteroid sulfotransferase is expressed in most ciliated, nonciliated, and basal airway cells, but not in mucus-secreting cells. Human phenol sulfotransferase is highly expressed and widely distributed in fetal lung [172, 178]. After birth, expression decreases and the distribution is restricted to the proximal airways.
Cellular Injury in Response to Toxicant Exposure
The respiratory system is a target for a wide range of toxic environmental contaminants. While the acute and chronic effects of a large number of these substances have been well characterized for the respiratory system of young adult mammals, there is significantly less known about the impact of these lung-targeted compounds on the developing and aged respiratory system. Recent studies have begun to identify mitochondria a target of many of these compounds and also highlight their role in the progression of cellular injury. This section summarizes what is currently known about three classes of these compounds, addressing first environmental tobacco smoke, a well-recognized lung toxicant mixture, bioactivated environmental contaminants, and oxidant gases, all of which have been shown to affect the developing and/or aged lung.
Environmental Tobacco Smoke
The health consequences of exposure to environmental tobacco smoke (ETS) among children are the ongoing subject of much public health concern. Animal studies are very valuable because the indirect effects of in utero ETS exposure can be separated from the direct effects of postnatal ETS exposure.
Exclusively in utero exposure to ETS has been shown to accelerate the developmental pattern of Clara cell secretory protein expression in the rat [179], suggesting a potential acceleration of airway epithelial differentiation in the lung. Whether this accelerated development is maintained after birth is unknown. In utero exposure does not increase cytochrome P450 gene expression unless it is combined with an early postnatal exposure [180].
Exclusively postnatal ETS exposure in rats did not alter Clara cell secretory protein expression [131]. Postnatal ETS exposure did, however, decrease cell kinetic activity in distal airways and increase cytochrome P450 1A1 protein distribution throughout the airway tree in postnatal ETS chronically exposed rats [131]. These changes were maintained for up to 100 days (with ongoing ETS exposure). Separate from the other compounds in ETS, nicotine exposure during gestation and lactation has been shown to increase CYP2A3 and CYP2B1 mRNA [181]. Acute postnatal exposure to ETS in juvenile ferrets has been shown to increase the ability of the lungs to metabolize (−)-trans-benzo[a]pyrene-7,8-dihydrodiol [182]. When slightly older rats (weanling age) are exposed to tobacco smoke, emphysematous changes are reported in their lungs [183]. Postnatal exposure to ETS has also been reported to affect the neurophysiologic responses of the lung. Chronic ETS exposure during the period of postnatal lung development in guinea pigs has been shown to increase lung C-fiber sensitivity [184]. In addition, ETS can increase the sensitivity of C-fiber-activated neurons in the nucleus tractus solitarii (NTS) of the central nervous system [185, 186].
A combination of in utero and postnatal exposure to ETS appears to have the greatest effect on developing lungs. Rats exposed to both in utero and postnatal ETS have decreased lung compliance, increased reactivity to methacholine, and an increase in the number of neuroendocrine cells per cm of basal lamina [187]. These changes are not seen in rats exposed to ETS in utero only or postnatal only. The increased airway hyperresponsiveness that is set up during postnatal exposure does not resolve even after an extensive period of no exposure to ETS [188]. In nonhuman primates, in utero plus postnatal exposure to ETS increases pulmonary adenyl cyclase activity [189]. Recent studies have focused on the effects that in utero exposure to ETS may have on the etiology of asthma. Early life exposures to ETS in the nonhuman primate enhance local Th2 immunity by impairing normal Th1 immune maturation [190]. In the mouse, in utero ETS exposure results in altered gene expression in adult animals [191] and exacerbating adult responses to allergen challenges [192]. In humans, in utero ETS exposure has been correlated to increased wheezing and increased doctor-diagnosed asthma by 2 years of age [193]. Whether in utero-induced alterations are the result of direct or indirect effects of ETS is unknown at this time.
ETS exposure is associated with increased GSH levels in the ELF; however, as mice age, response capacity is greatly reduced [96]. Unlike the developing lung, studies evaluating the effects of ETS on the adult and aged lung are limited; however, a large body of literature has found associations between cigarette smoking, premature aging, cancer, and COPD. Aged rats have a reduced ability to resist cigarette smoke-induced oxidative damage while still maintaining ability to activate PAHs to their carcinogenic form, shifting the balance to favor carcinogenesis with advanced age [90]. Senescent mice and senescence-accelerated mouse models have both been shown to have enhanced susceptibility to cigarette smoke [194–196]. Aging in the lung is believed to be accelerated by reactions between of components of cigarette smoke and extracellular matrix proteins to form covalent adducts that result in advanced glycation end products [116]. Advanced glycation end products (AGEs) play a role in aging and degenerative diseases due to their ability to crosslink proteins, especially structural proteins such as collagen [197]. Smoking has been shown to accelerate maturation of the fetal lung, impair lung growth, and accelerate age-related declines in FVC and FEV1 [198]. Reductions in FEV1 were also shown for cigar and pipe smokers who inhaled; however, non-inhaling pipe smokers had declines similar to nonsmokers [199].
Cigarette smoke constituents, such as phenols, aldehydes, and aromatic compounds, have been shown to accumulate in the mitochondria and disrupt ETC function and ATP production [200]. In a murine model for asthma, ETS exposure increases levels of ROS and lipid peroxides while decreasing mitochondrial enzymes [201]. In cell culture, cigarette smoke extracts (CSE) have been found to decrease membrane potential and ATP levels [202, 203]. In A549 cells, CSE increases intracellular ROS production; however, in cells the functional mitochondria (A549-rho0) have been depleted, suggesting that CSE increases mitochondrial ROS production. CSE also increases caspase-dependent apoptosis [202].
Bioactivated Compounds
The lungs of mammals are selectively injured by a host of chemically diverse agents including aromatic hydrocarbons, furans, halogenated ethylenes, and indoles [136]. Many of these agents target airway epithelium, especially Clara cells. In all cases, the metabolic activation of the chemically inert parent compounds has been demonstrated to be an important factor in selective lung injury. It is generally assumed that the Clara cell is most susceptible by virtue of the high expression of cytochrome P450 monooxygenases in this cell type. Despite extensive documentation of Clara cell susceptibility to P450-mediated cytotoxicants in the lungs of adults [106, 120, 121, 202, 204–211], little is known about the susceptibility of undifferentiated and developing cells in the neonate to these compounds. The studies that are available regarding neonates counterintuitively suggest that lower pulmonary P450 activity is associated with greater susceptibility to P450-activated toxicants [128, 138, 212–214]. In utero exposure to bioactivated compounds produces embryotoxic or teratogenic effects, including chromosomal aberrations [215–220]. The latter appears to be the case for a number of pro-carcinogens which when given to pregnant mothers produce Clara cell tumors in adult offspring [219, 221, 222].
The herbicide dichlobenil specifically injures olfactory nasal mucosa in fetal and neonatal mice just as it does in adult mice [223]. The toxicity of dichlobenil increases in neonatal mice with the development of Bowman’s glands. In fetal mice, there is more irreversible binding of 14C-dichlobenil in the nasal cavity when dichlobenil is given to the dam as opposed to injected directly into the fetus. This suggests that maternal metabolism of dichlobenil may be important in the fetus.
While the toxicity of dichlobenil increases with development of the target organ, this is not always true for the other bioactivated compounds in the developing lung. Neonatal rabbits are much more sensitive to the P450-bioactivated furan 4-ipomeanol as compared to adult rabbits [213]. Distal airway epithelium of neonatal rabbits is injured at doses that do not appear to affect the adult rabbit airway epithelium. This would seem to be a contradiction because development of the P450 system is a postnatal event, and in neonatal rabbits, P450 activity is very low. This phenomenon is not restricted to the rabbit. Studies have shown that neonatal mice are also more susceptible than adult mice to the Clara cell cytotoxicant naphthalene [212] and neonates of both rats and mice are more susceptible to 1-nitronaphthalene than are adult animals [138]. Both 4-ipomeanol [224] and naphthalene [225, 226] have been shown to alter mitochondrial morphology in nonciliated cells in the terminal bronchioles of mice. In the hours and days after injury caused by either 4-ipomeanol or naphthalene administration, increase in the mitochondrial swelling has been noticed, suggesting that mitochondria may be a target of naphthalene metabolites, although studies to directly investigate this have not been performed.
In vitro metabolism studies of neonatal and adult airways show that P450 activity is lower in neonatal mouse lung than it is in adult mouse lung [129]. Gender may also play a role in heightened postnatal sensitivity to pulmonary toxicants. Weanling male and female mice are reported to be more susceptible to 1,1-dichloroethylene-induced pulmonary injury than adult male mice, but not more susceptible than adult female mice [227]. The exact mechanisms of these increases in sensitivity of postnatal animals have yet to be clearly defined. In some cases, levels of injury positively correlate with specific P450 monooxygenase activity [227], while in other cases phase II enzyme activity may be key to increased susceptibility [148]. The mechanisms may also involve as yet undefined factors specific to differentiating cells such as the important role of mitochondria in cellular injury.
Oxidant Gases
Lung injury occurs when concentrations of inhaled oxidant gases exceeds the detoxification capacity of the lung. The pulmonary effects of oxidant gases are dependent on a number of factors, including the age at exposure; chemical properties of the gas, which include solubility and reactivity; and the effective dose which is in part dependent upon ventilatory rates, concentration, and duration of exposure [228–230].
In contrast to our understanding of P450-activated lung toxicants, the susceptibility of the lungs of postnatal animals to oxidant gases is much better understood. For the best-studied oxidant gas environments (hyperbaric oxygen, ozone and nitrogen dioxide), two fundamental characteristics have been defined. First, in general, postnatal animals, prior to weaning, are less susceptible to pulmonary injury than are adults. Second, exposure to oxidant gases retards postnatal maturation of the lung.
The tolerance of postnatal animals to hyperoxia appears to be species-specific [70, 231, 232] and is based on differences in (1) the ability of neonatal animals to elevate pulmonary antioxidant defense systems in response to hyperoxic stress [233–235], (2) the composition of lung polyunsaturated fatty acids [236], or (3) the presence of antioxidant compounds, including iron chelators [82]. A common factor appears to be the ability to increase the intracellular GSH pool and to upregulate the enzymes whose antioxidant functions depend on it, including SOD, catalase, GPx, and glucose 6-phosphate dehydrogenase [70, 232, 237–240]. Undernutrition and premature weaning have also been shown to alter susceptibility [82, 241, 242]. Pharmacologic intervention by administration of steroids (dexamethasone) or endotoxin attenuates neonatal susceptibility to hyperoxia but has a mixed effect on antioxidant enzyme activity (endotoxin elevates them, dexamethasone does not) [243, 244]. Hyperoxia has been shown to delay lung morphogenesis, including alveolarization and vascularization [245–248], and differentiation of Clara cells in postnatal rats [249]. Studies in mice have shown that the decreased alveolarization is due to arrested alveolar development that can be attributed to decreased respiratory control ratios and decreased O2 consumption, suggesting impaired mitochondrial function [250]. Treatment with retinoic acid does not prevent hyperoxia-induced alterations in alveolarization; however, it does result in later improvement in alveolarization [251, 252]. Excessive collagen deposition (fibrosis) that is associated with the decreased alveolar and capillary development is preceded by an increase in connective tissue growth factor (CTGF) in neonatal rats [253]. Despite alterations in lung development, neonatal rats have been reported to survive hyperoxia longer than adult rats. This may partially be due to the fact that in neonatal rats, there is a delay in pulmonary neutrophil influx [254]. Compared to adult rats, neonatal rats have fewer overall lung tissue neutrophils, even though they have higher levels of neutrophils in bronchoalveolar lavage. This suggests that neonatal rats retain fewer neutrophils than adult rats [254]. When neonatal rats exposed to hyperoxia were treated with antibodies to cytokine-induced neutrophil chemoattractant-1 (CINC-1) to block neutrophil influx, they had increased lung compliance and no change in alveolar volume or surface density as compared to control antibody-treated neonates [255]. In addition, blocking neutrophil influx reduces DNA damage in the neonatal lung [256]. The retardation of alveolar development in neonatal lung may also be related to the timing of the hyperoxia and subsequent exposure to leukotrienes [257] or a reported increase in the number of apoptotic cells in the lungs of hyperoxia-exposed neonates [258].
Rats exposed to hyperoxia during the first 7 days of life have shown increased volume density of mitochondria in both ciliated and Clara cells as well as reduced nuclear density of bronchiolar epithelial cells immediately after exposure [259]. However, recovery in filtered air resulted in a reversal of these changes. In adult rats exposed to 70 % oxygen, mitochondria have shown increased ROS in the matrix, suggesting that an increase in the oxidative burden is mitochondrially derived [260]. After exposure, an increased number of cells entered a premature senescent state, which may be due to the increased ROS production resulting in mtDNA damage and mitochondrial dysfunction.
For some parameters, neonates appear to be less susceptible to inhaled oxidant gases due to fewer alterations in pulmonary enzymes and markedly decreased cellular injury in the central acinus as compared to adults [261–265]. Weaning appears to be the critical time point for changes in responsiveness. Pre-weanling animals are much less sensitive than post-weaning animals [262, 264, 265]. As in hyperoxia, ozone exposure reduces the postnatal morphogenesis of the gas-exchange area [266], impairs bronchiolar formation [267], and retards the differentiation of the mucociliary apparatus of proximal airways [268]. Nonhuman primates exposed to cyclic episodes of ozone during the first 6 months of life were found to have four fewer non-alveolarized airway generations, hyperplastic bronchiolar epithelium, and altered smooth muscle bundle orientation in terminal and respiratory bronchioles compared to filtered air-exposed control animals [269] and hyperinnervation of the pulmonary epithelium [270]. The molecular mechanism behind the abnormal development of distal conducting airways in animals exposed to ozone may be related to the depletion of perlecan in the basement membrane zone [271]. Perlecan is a proteoglycan responsible for many functions, in particular, regulation of growth factor trafficking between cells of the epithelial-mesenchymal unit [272, 273]. Ozone-induced depletion of perlecan from the basement membrane zone in trachea has been shown to be associated with altered regulation of FGF-2 signaling [271, 274]. Depletion of perlecan would also effect regulation of the other growth factors that bind to perlecan which also include FGF-1, FGF-7, PDGF, hepatocyte growth factor, and heparin-binding EGF, VEGF, and TGF-β [275]. The functional consequences of deregulation of these collective molecules are significant since they are the basis for much of the cell-cell interactions in the epithelial-mesenchymal trophic unit responsible for development of the airway. The dysregulation of the epithelial-mesenchymal trophic unit may play a role in explaining the epidemiological findings regarding children who are exposed to long-term outdoor air pollution. In Southern California, an association has been found between long-term exposure of children to outdoor air pollution and deficits in lung function as these children become adolescents and young adults [276–279]. Children growing up in Mexico City, which has very high levels of ozone and other air pollutants, have respiratory abnormalities such as hyperinflation and increased interstitial markings of the lung [280–282].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


