Wall constituent
Changes
Myocytes
Hypertrophy, lengthening
Reexpression of fetal gene program
Altered excitation–contraction coupling
Impaired energy metabolism
Defective myofibrillar content and function
Apoptosis, necrosis
Vasculature
Endothelial dysfunction
Intimal thickening
Smooth myocyte hyperplasia
Rarefication of capillaries
Interstitium
Activation of matrix metallopeptidases
Increased collagen synthesis, fibrosis
Collagen isoform shift
The cardiomyocyte is the major cell involved in the pathological remodeling. Other implicated components include fibroblasts as well as the interstitium with collagen fibers and coronary vasculature. Relevant processes encompass ischemia and subsequent cell apoptosis and necrosis. Clinical manifestations comprise variations of size, shape, and function.
Processes occurring in ventricular remodeling include [208]:
1.
Cardiomyocyte lengthening, hypertrophy, and loss
2.
Inflammation
3.
Excessive accumulation of collagen in the cardiac interstitium
4.
Ventricular wall thinning and dilation and reshaping of the left ventricle that becomes more spherical
5.
After myocardial infarction, reabsorption of necrotic tissue, scar formation, and continued infarct expansion rather than extension
The neurohormonal context implicates especially the sympathetic nervous system and the renin–angiotensin axis as well as oxidative stress that describes the imbalance between production and/or activity of oxygen-derived free radicals and those of antioxidant factors. Various substances are involved, such as noradrenaline, aldosterone, renin, angiotensin-2, potent vasoconstrictor endothelin, atrial natriuretic peptide, nitric oxide, and cytokines (tumor necrosis factor TNFSF1 and interleukins).
The myocardium consists of myocytes tethered and supported by a connective tissue composed largely of fibrillar collagen. When cardiomyocytes stretch, the local activity of noradrenaline, angiotensin-2, and endothelin rises, thereby provoking adverse myocyte hypertrophy. In addition, fibroblasts proliferate. They synthesize and degrade the extracellular matrix. Moreover, elevated activation of aldosterone and cytokines can also stimulate collagen synthesis, thus engendering fibrosis. On the other hand, collagenase present as an inactive proenzyme in the cardiac wall can be activated after myocardial injury [208].
Cardiac dilation can happen without hypertrophy. Myocardial stress then increases and can generate further dilation of the heart.
Adverse cardiac remodeling causes a progressive worsening of cardiac function and then heart failure. Hence, therapy is aimed at slowing and even reversing maladaptive remodeling, thus enhancing cardiac function (end-diastolic and end-systolic volume and ejection fraction). The antineuroendocrine therapy with angiotensin-converting enzyme inhibitors and β-adrenergic blockers improve ejection fraction and ventricular volumes [209].
Cardiac-impaired structure with inflammation, oxidative stress, and remodeling (fibrosis) and ventricular dysfunction is also observed during the development of cardiomyopathies.
3.1 Cardiac Fibrosis
Fibrosis affects the architecture and structure of the heart. It arises during cardiac remodeling. Fibrosis is the feature of numerous types of cardiomyopathies (e.g., hypertrophic and inflammatory cardiomyopathies). Myocardial fibrosis can engender diastolic (abnormal left ventricular filling and reduced compliance and elevated diastolic pressure) and/or systolic dysfunction and ultimately heart failure. Inflammation and fibrosis are correlated with diastolic dysfunction in heart failure with a preserved ejection fraction.
Fibrosis is mainly engendered by collagen-secreting myofibroblasts, which originate not only from cardiofibroblasts but also from other cell sources, such as endothelial and circulating cells.
Fibroblasts are pleiomorphic and pleiotropic cells of connective tissues responsive to numerous profibrotic factors. Conversely, cardiac fibrosis triggers endothelial–mesenchymal transition that contributes to about 30 % of activated fibroblasts.
Cardiofibroblasts ensure the maintenance of the extracellular matrix, as they modulate the production of matrix constituents (e.g., collagen-1 and collagen-3 to collagen-6) and degraders (e.g., matrix metalloproteinases). They also secrete growth factors and cytokines that exert auto- and paracrine effects on cell fate (e.g., proliferation and apoptosis). Under pathological conditions, in response to proinflammatory cytokines (e.g., TNFSF1, IL1, IL6, and TGFβ), cardiofibroblasts have a higher proliferation, migration, and differentiation rate and secrete more collagens, MMPS, and cytokines. These agents contribute to the development of perivascular and interstitial fibrosis. Cardiofibroblasts contribute to cardiac fibrosis via many synergistic factors (e.g., ATn2, ET1, TGFβ, and CTGF).
Collagen production and collagen fiber formation involve multiple stages. Following assembly into a triple helix structure, procollagen is transported in specialized CoP2+ vesicles and secreted into the extracellular space. Procollagen is then processed by peptidases that cleave the N- and C-propeptide-forming collagen that self-assembles into fibrils.1
For example, once secreted, procollagen-1 is converted to mature collagen-1 by adamlysin with thrombospondin motifs ADAMTS2 that excises the N-terminal propeptide to generate 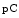 collagen-1 and by bone morphogenic protein BMP1 that excises the C-terminal propeptide to produce
collagen-1 and by bone morphogenic protein BMP1 that excises the C-terminal propeptide to produce 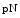 collagen-1 [211]. The collagen on primary murine dermal fibroblast surfaces comprises four forms: procollagen-1,
collagen-1 [211]. The collagen on primary murine dermal fibroblast surfaces comprises four forms: procollagen-1, 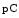 and
and 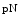 collagen-1, and collagen 1. In some cases, propeptides are retained following incorporation into fibrils.
collagen-1, and collagen 1. In some cases, propeptides are retained following incorporation into fibrils.
collagen-1 and by bone morphogenic protein BMP1 that excises the C-terminal propeptide to produce collagen-1 [211]. The collagen on primary murine dermal fibroblast surfaces comprises four forms: procollagen-1, and collagen-1, and collagen 1. In some cases, propeptides are retained following incorporation into fibrils.Procollagen lysine (1,2)-oxoglutarate 5-dioxygenase PLOD1 (or lysyl hydroxylase LH1) converts lysine into hydroxylysine and lysyl oxidase (LOx) catalyses cross-linking of collagen fibrils and formation of collagen fibers via oxidative deamination of lysine and hydroxylysine.
The matricellular proteins,2 secreted protein acidic and rich in cysteine (SPARC)3 and secreted phosphoprotein 1 (SPP1),4 are major contributors to posttranslational modification, stabilization, and deposition of collagen in the heart.
Collagen-binding SPARC is produced by cardiofibroblasts. It is required for aggregation of collagen fibrils (diameter 60–70 nm), an intermediate step in collagen fiber assembly [212]. It mediates the tethering of procollagen-1 to cells as well as its subsequent processing and incorporation into the extracellular matrix. In the absence of SPARC, the amount of cell surface-associated collagen and the proportion of total collagen-1 without propeptides on SPARC-null fibroblasts increase [213].
Secreted acidic phosphoglycoprotein SPP1 functions in tissue repair, matrix organization, and collagen fibrillogenesis. It intervenes in the development of collagen-1-induced cardiac fibrosis and dysfunction in human dilated cardiomyopathy [214]. Collagen-1 upregulates SPP1 expression, but not fibronectin, laminin, or vitronectin [215]. It is detected in atherosclerotic plaques. It connects to collagen-1 to -3 and -5 synthesized by vascular smooth myocytes as well as collagen-4 in basement membranes [216]. It is targeted by widespread intra- and extracellular Ca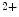 -dependent tissue transglutaminase that catalyzes the formation of high-molecular-mass complexes of its proteic substrates, as it cross-links glutamine and lysine residues [216]. As is SPARC, SPP1 is linked to fibrosis. In SPP1
-dependent tissue transglutaminase that catalyzes the formation of high-molecular-mass complexes of its proteic substrates, as it cross-links glutamine and lysine residues [216]. As is SPARC, SPP1 is linked to fibrosis. In SPP1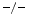 mice, angiotensin-2-induced myocardial fibrosis is hindered. On the other hand, the cytokine IL1β potentiates angiotensin-2-induced SPP1 expression by fibroblasts. Macrophages and neutrophils also produce SPP1.
mice, angiotensin-2-induced myocardial fibrosis is hindered. On the other hand, the cytokine IL1β potentiates angiotensin-2-induced SPP1 expression by fibroblasts. Macrophages and neutrophils also produce SPP1.
-dependent tissue transglutaminase that catalyzes the formation of high-molecular-mass complexes of its proteic substrates, as it cross-links glutamine and lysine residues [216]. As is SPARC, SPP1 is linked to fibrosis. In SPP1 mice, angiotensin-2-induced myocardial fibrosis is hindered. On the other hand, the cytokine IL1β potentiates angiotensin-2-induced SPP1 expression by fibroblasts. Macrophages and neutrophils also produce SPP1.3.1.1 Differentiation of Cardiofibroblasts into Myofibroblasts
Fibroblasts originated from diverse organs are heterogeneous according to size, shape, proliferative capacity, growth factor production rate, and collagen secretion rate.
Fibroblasts represent the most numerous cardiac cell type. Myofibroblasts appear in pathological conditions. Fibroblast differentiation is caused by the cytokine transforming growth factor-β1 (TGFβ-1) produced by inflammatory cells and mechanical stress.
Differentiation of cardiac fibroblasts into myofibroblasts supports collagen deposition and connective tissue construction during myocardial remodeling. It is also involved in augmented production of angiotensin by angiotensin convertase ACE1.
Differentiation of cardiofibroblasts consists of two stages [217]:
1.
Differentiation to protomyofibroblasts with 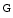 actin polymerization into
actin polymerization into 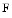 actin stress fibers
actin stress fibers
actin polymerization into actin stress fibers2.
Further differentiation into myofibroblasts in which stress fibers are decorated with α-smooth muscle actin (SMA)
Polymerization of stress fibers prevents sequestration of myocardin-related transcription factors in the cytosol by globular actin. In addition, TGFβ1 and the RoCK kinase increase the 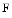 actin/
actin/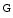 actin ratio, thereby favoring MRTF translocation to the nucleus. Inside the nucleus, MRTFs support activation of α-SMA synthesis [217]. Suppression of α-SMA diminishes arrhythmias in cardiomyocytes cocultured with cardiac myofibroblasts.
actin ratio, thereby favoring MRTF translocation to the nucleus. Inside the nucleus, MRTFs support activation of α-SMA synthesis [217]. Suppression of α-SMA diminishes arrhythmias in cardiomyocytes cocultured with cardiac myofibroblasts.
actin/actin ratio, thereby favoring MRTF translocation to the nucleus. Inside the nucleus, MRTFs support activation of α-SMA synthesis [217]. Suppression of α-SMA diminishes arrhythmias in cardiomyocytes cocultured with cardiac myofibroblasts.Adult rat cardiac fibroblasts cultured on a stiff plastic substratum spontaneously differentiate to proliferating myofibroblasts characterized by stress fibers connected to α-SMA [217].
Fibroblasts in collagen matrices do not exhibit any contraction, whereas quiescent and proliferating myofibroblasts contract. Developed cell tension can provoke TGFβ1 production. Transforming growth factor-β1 promotes differentiation into α-SMA+ quiescent myofibroblasts via the Tβ R1 receptor [217].
Fibroblasts produce small amounts of collagen, but high levels of interleukin-10 [217]. Quiescent myofibroblasts synthesize collagen, tissue inhibitor of metalloproteinases TIMP1, and the CCL2 chemokine at high concentrations. The transcriptome shows that different gene networks are activation in myofibroblasts (e.g., paxilin and PAK) and quiescent myofibroblasts (cell cycle regulators) [217]. In particular, MRTFa required for activation of myofibroblast differentiation is distinctly regulated in fibroblasts and quiescent and proliferating myofibroblasts. The expression of MRTFa is low in quiescent myofibroblasts [217].
Dedifferentiation of proliferating myofibroblasts associated with stress fiber depolymerization, but not of quiescent myofibroblasts, results from Tβ R1 receptor inhibition and mechanical strain reduction [217]. Loss of strain in the culture medium also reduces α-SMA expression in noncardiacmyofibroblasts and collagen-1 production.
3.1.2 Profibrotic Signaling
Cardiac fibrosis is characterized by fibroblast proliferation, migration, and differentiation and extracellular matrix turnover.
3.1.2.1 Selective Atrial Fibrosis by TGF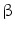
Selective atrial fibrosis relies on TGF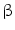 . The latter is an important activator of chemokines, thereby attracting monocytes into the cardiac wall. When the TGFβ production increases to a similar level in both atrio- and ventriculomyocytes, only the atria respond to TGFβ by interstitial fibrosis, although levels of signaling mediators (Tβ Rs, SMADs, and AP1) are similar in atria and ventricles [218]. Atria are indeed characterized by an enhanced receptor binding and receptor kinase activity and hence phosphorylation of SMAD2 and SMAD3 as well as reduced synthesis of inhibitory SMAD7 with respect to ventricles [218].
. The latter is an important activator of chemokines, thereby attracting monocytes into the cardiac wall. When the TGFβ production increases to a similar level in both atrio- and ventriculomyocytes, only the atria respond to TGFβ by interstitial fibrosis, although levels of signaling mediators (Tβ Rs, SMADs, and AP1) are similar in atria and ventricles [218]. Atria are indeed characterized by an enhanced receptor binding and receptor kinase activity and hence phosphorylation of SMAD2 and SMAD3 as well as reduced synthesis of inhibitory SMAD7 with respect to ventricles [218].
. The latter is an important activator of chemokines, thereby attracting monocytes into the cardiac wall. When the TGFβ production increases to a similar level in both atrio- and ventriculomyocytes, only the atria respond to TGFβ by interstitial fibrosis, although levels of signaling mediators (Tβ Rs, SMADs, and AP1) are similar in atria and ventricles [218]. Atria are indeed characterized by an enhanced receptor binding and receptor kinase activity and hence phosphorylation of SMAD2 and SMAD3 as well as reduced synthesis of inhibitory SMAD7 with respect to ventricles [218].Improved TGFβ1–Tβ R1/2 connection is favored by increased expression of connective tissue growth factor, Tβ R3 (endoglin), and atrial natriuretic peptide [219]. The enhanced activation of Tβ R together with lowered SMAD7 expression increase receptor-regulated SMADs and their phosphorylation.
Receptor-regulated rSMADs subsequently complex with SMAD4, move into the nucleus, and bind to target genes. Many genes are differentially expressed in atria and ventricles, among which those that encode mediators of fibrosis (e.g., collagen and other matrix proteins, [218]). Other AP1-regulated genes (e.g., COL1, FN1, PAI1, and LOX) are upregulated in the atria, but not in the ventricle.
3.1.2.2 Platelet-Derived Growth Factor-D
Platelet-derived growth factor-D and its receptor PDGFRβ, which is primarily expressed by fibroblasts, are implicated in the development of cardiac fibrosis [220]. The profibrogenic action of PDGFd is mediated by activation of the TGFβ1 pathway. Moreover, TGFβ1 exerts a positive feedback on PDGFd synthesis.
The PDGFd subtype augments [220]:
Cardiofibroblast proliferation and myofibroblast differentiation
Synthesis and secretion of collagen-1, matrix metallopeptidases (MMP1–MMP2 and MMP9) and their inhibitors (TIMP1–TIMP2)
TGFβ1
3.1.2.3 Vascular Endothelial Growth Factor-C (VEGFc)
The vascular endothelial growth factor subtype VEGFc is involved in lymphangiogenesis (Vol. 5, Chap. 10. Vasculature Growth). In addition, the VEGFc–VEGFR3 axis participates in fibrogenesis during cardiac repair. The VEGFR3 receptor is expressed not only in lymph ducts but also in myofibroblasts.
The VEGFc isoform activates the TGFβ1 pathway and primes ERK phosphorylation, promotes myofibroblast proliferation and migration as well as production of collagen-1 and -3, matrix metallopeptidases MMP2 and MMP9, and tissue inhibitor of metallopeptidases TIMP1 and TIMP2 [221].
3.1.2.4 Tissue Inhibitors of Metallopeptidases
Tissue inhibitors of metallopeptidases and hence the TIMP–MMP balance participate in adverse myocardial remodeling (hypertrophy and fibrosis) as well as termination of the immune response [222]. Cardiac pressure overload provokes severe cardiac fibrosis and hypertrophy in mice lacking TIMP2 and TIMP3, but not in TIMP4 [223].
Among tissue inhibitors of matrix metallopeptidases, antihypertrophic TIMP2 and antiinflammatory, antifibrotic TIMP3 differ in their action in adverse cardiac remodeling engendered by the fibrogenic and hypertrophic messenger angiotensin-2 [223]. In response to angiotensin-2 infusion during 2 weeks, TIMP2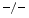 , TIMP3
, TIMP3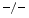 , and wild-type (WT) mice still have a preserved left ventricular ejection fraction. However, both TIMP2
, and wild-type (WT) mice still have a preserved left ventricular ejection fraction. However, both TIMP2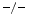 and TIMP3
and TIMP3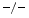 mice are characterized by left ventricular diastolic dysfunction. Furthermore, TIMP2
mice are characterized by left ventricular diastolic dysfunction. Furthermore, TIMP2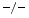 mice exhibit an impaired relaxation and a larger degree of myocardial hypertrophy without fibrosis, whereas TIMP3
mice exhibit an impaired relaxation and a larger degree of myocardial hypertrophy without fibrosis, whereas TIMP3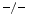 mice have an increased left ventricular stiffness and inflammation associated with an excessive fibrosis without hypertrophy.
mice have an increased left ventricular stiffness and inflammation associated with an excessive fibrosis without hypertrophy.
, TIMP3, and wild-type (WT) mice still have a preserved left ventricular ejection fraction. However, both TIMP2 and TIMP3 mice are characterized by left ventricular diastolic dysfunction. Furthermore, TIMP2 mice exhibit an impaired relaxation and a larger degree of myocardial hypertrophy without fibrosis, whereas TIMP3 mice have an increased left ventricular stiffness and inflammation associated with an excessive fibrosis without hypertrophy.Adult wild-type mouse cardiomyocytes undergo hypertrophy upon angiotensin-2 exposure only when cocultured with cardiofibroblasts, except in TIMP3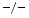 cardiomyocytes [223]. Inflammation and fibrosis characterize cultures of quiescent and cyclically stretched cardiofibroblasts of TIMP3
cardiomyocytes [223]. Inflammation and fibrosis characterize cultures of quiescent and cyclically stretched cardiofibroblasts of TIMP3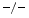 hearts. Fibrosis results from collagen deposition by the matrix proteins SPARC and SPP1 (Sect. 3.1). On the other hand, attenuated collagen deposition in TIMP2
hearts. Fibrosis results from collagen deposition by the matrix proteins SPARC and SPP1 (Sect. 3.1). On the other hand, attenuated collagen deposition in TIMP2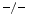 hearts exposed to angiotensin-2 is associated with reduced levels of the cross-linking enzymes lysyl oxidase and procollagen-lysine (1,2)-oxoglutarate 5-dioxygenase PLOD1 [223].
hearts exposed to angiotensin-2 is associated with reduced levels of the cross-linking enzymes lysyl oxidase and procollagen-lysine (1,2)-oxoglutarate 5-dioxygenase PLOD1 [223].
cardiomyocytes [223]. Inflammation and fibrosis characterize cultures of quiescent and cyclically stretched cardiofibroblasts of TIMP3 hearts. Fibrosis results from collagen deposition by the matrix proteins SPARC and SPP1 (Sect. 3.1). On the other hand, attenuated collagen deposition in TIMP2 hearts exposed to angiotensin-2 is associated with reduced levels of the cross-linking enzymes lysyl oxidase and procollagen-lysine (1,2)-oxoglutarate 5-dioxygenase PLOD1 [223].Expression of TIMP1 increases upon angiotensin-2 infusion. Elevated TIMP1 levels are frequently linked to fibrosis. However, its augmentation in TIMP2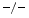 angiotensin-2-subjected hearts does not cause marked fibrosis despite an elevated collagen mRNA level with respect to excessive fibrosis in TIMP3
angiotensin-2-subjected hearts does not cause marked fibrosis despite an elevated collagen mRNA level with respect to excessive fibrosis in TIMP3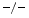 hearts without increased synthesis of collagen. In cultured quiescent mouse cardiofibroblasts, angiotensin-2 usually induces collagen production. However, TIMP2 raises collagen production by cardiofibroblasts and TIMP3 provokes cardiofibroblast apoptosis [223].
hearts without increased synthesis of collagen. In cultured quiescent mouse cardiofibroblasts, angiotensin-2 usually induces collagen production. However, TIMP2 raises collagen production by cardiofibroblasts and TIMP3 provokes cardiofibroblast apoptosis [223].
angiotensin-2-subjected hearts does not cause marked fibrosis despite an elevated collagen mRNA level with respect to excessive fibrosis in TIMP3 hearts without increased synthesis of collagen. In cultured quiescent mouse cardiofibroblasts, angiotensin-2 usually induces collagen production. However, TIMP2 raises collagen production by cardiofibroblasts and TIMP3 provokes cardiofibroblast apoptosis [223].TIMP2 is the main inhibitor of MMP2 that degrades inflammatory and profibrotic cytokines, thereby favoring inflammation and fibrosis. Both LOx and PLOD1 elicit collagen cross-linking and fibre formation; their level lowers in TIMP2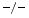 angiotensin-2-subjected hearts [223].
angiotensin-2-subjected hearts [223].
angiotensin-2-subjected hearts [223].The TIMP3 subtype is the single TIMP species that can inhibit ADAM17 sheddase, hence reducing activity of proinflammatory cytokines (e.g., TNFSF1 and IL6), as well as HER ligands (TGFα, amphiregulin, and HBEGF) via P38MAPK, ERK1 and ERK2 pathways. In addition, it attenuates action of profibrotic TGFβ and of collagen cross-linking stimulators SPARC and SPP1, thereby preventing the posttranslational processing and deposition of collagen fibers and hence the perivascular and interstitial myocardial fibrosis.
3.1.2.5 Eicosanoids and Their Inhibitors
Eicosanoids are potent modulators of immunity. They derive from arachidonic acid processed by cyclooxygenases, lipoxygenases, and cytochrome-P450 epoxygenases. The latter generates cardioprotective epoxyeicosatrienoic acids (EET) that act as auto- and paracrine effectors. However, EETs are further metabolized by soluble epoxide hydrolase into corresponding dihydroxyeicosatrienoic acids (DHET).
Myocardial infarction causes cardiomyocyte necrosis and adverse cardiac remodeling with fibrosis. Several soluble epoxide hydrolase inhibitors of postinfarction cardiac remodeling improve the cardiac function, as they impede structural (cardiac fibrosis and adverse hypertrophy) and electrical remodeling [224]. The proliferative capacity of different populations of cardiofibroblasts as well as recruitment of fibroblasts originated from circulating bone marrow-derived cells assisted by chemokines (e.g., CCL2) drop.
3.1.3 Fibrosis and Between-Cell Crosstalk
Intercellular communication between several cardiac and recruited cell types is involved in fibrosis, in particular crosstalk between cardiac myocytes and fibroblasts.
Fibroblasts are a regulating and secreting cell type that is not only implicated in maintaining the extracellular matrix of myocytes but also in their fate. In particuler, the cytokines TNFSF1 and IL6 secreted from fibroblasts can cause cardiomyocyte hypertrophy and may modulate their electrophysiological behavior and contractility.
Moreover, cellular communication can change the cardiac cell phenotype, especially during fibrosis. An endothelial–mesenchymal transition generates fibroblasts using Snail transcription factor in endotheliocytes.
Furthermore, endotheliocytes can release connective tissue growth factor (CTGF) that provokes fibrosis, as it engenders the change from quiescent fibroblasts to secreting myofibroblasts.
3.2 Maladaptive Myocardial Hypertrophy
Maladaptive, or adverse, myocardial remodeling occurs in response to sustained hypertension, myocardial infarction, and exposure to infectious or cardiotoxic agents. Hypertensive heart disease corresponds to left ventricular hypertrophy caused by prolonged arterial hypertension. Although the left ventricle is a major target, the right ventricle is involved in structural congenital heart defects with pulmonary valve stenosis, pulmonary arterial hypertension, in addition to cardiomyopathy and ischemic heart disease.
Inflammation (Vol. 5, Chap. 11. Tissue Growth, Repair, and Remodeling) and fibrosis that occur during myocardial remodeling contribute to cardiac dysfunction. Fibrosis corresponds to an excessive formation of fibrous connective tissue as a reactive process. Aggravation results from a defective neurohumoral control ( sympathetic nervous regulation and renin–angiotensin–aldosterone signaling; Vol. 8, Chap. 4. Hypertension).
Hypertensive myocardium is characterized by structural changes of cardiomyocyte and matrix, such as cardiomyocyte hypertrophy and excessive apoptosis, accumulation of interstitial and perivascular collagen fibers, and disruption of endomysial and perimysial collagen network.
This pathological remodeling facilitates the development of heart failure. Progression to cardiac failure is exhibited by increase in relative lung and right ventricular weights, cardiac function disorders, and overexpression of type-A and -B natriuretic peptides.
3.2.1 Cardiac Cell Interactions in Maladaptive Cardiac Hypertrophy
Cardiac hypertrophy is related to two distinct biological process, compensatory (physiological) and deleterious (pathophysiological) cardiac hypertrophy. The molecular mechanisms underlying different forms of hypertrophy depend on the type of stress and subsequent signaling. The PI3K–PKB–TOR cascade is preferentially activated in exercise- and IGF1-dependent adaptative hypertrophy (Vol. 5, Chap. 5. Cardiomyocytes). On the other hand, activation of the PP3 and MAPK pathways is related to adverse cardiac hypertrophy in response to pressure overload.
In adverse cardiac hypertrophy, an increased myocardial mass, fibroblast activation, deposition of an extracellular matrix, and neovascularization are slower than after acute injury. Maladaptive cardiac hypertrophy is characterized by an elevation of the cardiomyocyte size, fibrosis, and expression of a fetal gene program in affected myocytes.
Myofibroblasts contribute to the cardiac response to high blood pressure. M2 macrophages are also involved in cardiac hypertrophy.
Cardiac hypertrophy is associated with an augmented blood vessel density. In heart hypertrophy due to pressure overload, the HIF1–VEGF signaling enables the maintenance of the capillary density.
MicroRNA-155 produced in cardiomacrophages and T cells induces cardiac hypertrophy and inflammation via SOCS1 factor [74].
The GSK3β kinase regulates the canonical Wnt signaling. Overexpression of GSK3β attenuates cardiac hypertrophy and its inhibition augments hypertrophy in response to hypertrophic stimuli [35].
Nitric oxide has an antihypertrophic role in cardiomyocytes, as it impedes the PP3–NFAT pathway upon an elevated cGMP–PKG1 activity and/or reduces effect of cytoskeletal LIM protein [64]. In addition, subsequently to angiogenic stimulation, NO promotes the degradation of regulator of G-protein signaling RGS4 that induces cardiomyocyte hypertrophy via the Gβγ–PI3Kγ–PKB–TORC1 pathway.
Multiple signaling pathways operate in cardiomyocytes and leukocytes in hypertensive heart disease (Tables 3.2 and 3.3).
Table 3.2
Signaling pathways associated with cardiomyocyte–leukocyte communication in hypertensive heart disease (Part 1; Source: [69])
Signaling axes in cardiomyocytes | Mediator effect |
|---|---|
mtDNA–TLR9–MyD88–NFκB | IL1β/6 Macrophage recruitment |
TLR4–TOR–NFκB | TNFSF1, IL1β/6 Macrophage recruitment |
ATn2–HSP70–TLR4 | CCL2 Macrophage recruitment |
LPS–TLR4–PI3Kγ | HMGB1 |
βAR + IL1βR–cAMP–PKA | IL6 Macrophage activation |
TLR4–P38MAPK/ERK—NFκB | TNFSF1 Mastocyte recruitment |
Table 3.3
Signaling pathways associated with cardiomyocyte–leukocyte communication in hypertensive heart disease (Part 2; Source: [69])
Signaling axes in cardiomyocytes | Mediator effect |
|---|---|
TNFSF1–NOx | IL1β/6 Macrophage activation |
TNFSF11–TNFRSF11a–TRAF2/6–PLC–PKC–NFκB | TNFSF1, IL1α/β Macrophage activation |
IL10–JaK–STAT3–NFκB | TNFSF1 Inhibition of monocyte recruitment |
EMMPRIn–Itg via PI3K–PKB–IKK–NFκB and MAP2K7–JNK–AP1 | IL18 Macrophage activation |
IL18–MyD88–IRAK4–TRAF6–JNK–SP1 | EMMPRIn MMP2 secretion by macrophage |
Hypertension affects cardiomyocyte mitochondria. During pressure overload, injured mitochondria that escape mitophagy release their DNA content. Mitochondrial DNA serves as alarmin (or danger-associated molecular pattern protein [DAMP]) recognized intracellularly by TLR9 on endosomes. The latter signals via MyD88 and primes the production of leukocyte recruiting factors such as IL6 [69].
Stressed cardiomyocytes also abundantly release heat shock protein HSP70 in the extracellular medium in response to angiotensin-2. The HSP70 signal binds to TLR4 on the membrane of surrounding cardiomyocytes, thereby initiating proinflammatory pathways [69].
The overexpressed TOR kinase hampers TLR4 signaling and secretion of TNFSF1, IL1β, and IL6 in cardiomyocytes [69]. Conversely, PI3Kγ promotes TLR4 signaling. This kinase is also an effector of β-adrenergic receptor that is constitutively engaged in pressure-overloaded hearts due to high levels of circulating catecholamines. In addition, the crosstalk between the β AR and IL1β cascades via the cAMP–PKA pathway provokes IL6 secretion [69].
In cardiomyocytes stimulated by lipopolysaccharides targeting TLR4 also engaged by pressure overload-related DAMPs, TNFSF1 expression upregulation involves P38MAPK and ERK kinases, and subsequently activated NFκB. This pathway is supported by phospholipase-Cγ and histone deacetylase and impeded by NO-induced Ca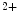 release, the JNK1–Fos axis, MAPK phosphatase MKP1, and GSK [69].
release, the JNK1–Fos axis, MAPK phosphatase MKP1, and GSK [69].
release, the JNK1–Fos axis, MAPK phosphatase MKP1, and GSK [69].Cardiomyocyte-derived TNFSF1 initiates an autocrine signaling involving the NADPH oxidases Nox2 and Nox4 that ensure a sustained secretion of inflammatory mediators. The TNFSF1 agent can control matrix remodeling indirectly by attracting leukocytes and directly by promoting myocyte apoptosis and activation of matrix metallopeptidases [69].
Another member of the TNF superfamily, TNFSF11 is released by cardiomyocytes in response to angiotensin-2 stimulation and recognized by the cardiomyocyte TNFRSF11a receptor. The latter is coupled to the TRAF2/6–PLC–PKC cascade that can lead to the nuclear translocation of NFκB [69].
In pressure overloaded, cardiomyocytes can signal to leukocytes by upregulating basigin (Bsg)5 due to high levels of catecholamines and reactive oxygen species [69]. Basigin connects to integrins on surrounding cardiomyocytes, thereby triggering the Rac1-dependent PI3K–PKB–IKK–NFκB and MAP2K7–JNK–AP1 axes and subsequently synthesizing proinflammatory cytokines (mainly IL18). Conversely, IL18 stimulates the BSG gene transcription in cardiomyocytes via the MyD88–IRAK4–TRAF6–JNK–SP1 pathway (positive feedback), thereby favoring MMP expression in cardiomyocytes and monocytes [69].
Termination of pressure overload-dependent inflammation relies on IL10 that tethers to IL10R and signals via P38MAPK and the JaK–STAT3 pathway to antagonize NFκB activation and TNFSF1 generation [69].
3.2.2 Metabolic Remodeling
The dysfunctional heart is characterized early by disturbed metabolism and impaired protein synthesis. Metabolic changes prime and perpetuate cardiac structural remodeling. In addition, nutrients (e.g., Ca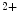 , Mg
, Mg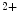 , Zn
, Zn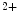 , Se
, Se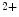 , and vitamin-D) can contribute to myocardial remodeling.
, and vitamin-D) can contribute to myocardial remodeling.
, Mg, Zn, Se, and vitamin-D) can contribute to myocardial remodeling. Angiotensin-2 responsible for cardiomyocyte maladaptive hypertrophy, interstitial fibrosis, and apoptosis promotes inflammation. Angiotensin-2 represses fatty acid oxidation. This effect is mediated by enhanced synthesis of tumor-necrosis factor-α.
3.2.2.1 Cardiac Energetics
The energy provider adenosine triphosphate (ATP) is required for both cell viability and myocardial pump activity. Cardiomyocytes are characterized by large rates of ATP use and synthesis, mainly by fatty acid oxidation in mitochondria.
At rest, fatty acid oxidation covers more than 70 % of energy need [225]. The remainder comes from oxidation of carbohydrates, principally glucose. In the healthy heart, selection among lipids and carbohydrates is governed by plasma substrate availability and hormonal regulation (insulin and catecholamines).
When heart activity rises, additional sources of ATP synthesis, such as glycogenolysis, glycolysis, and phosphotransferase reactions catalyzed by creatine kinase and adenylate kinase, are involved. Production of ATP by phosphotransferase reactions owing to creatine kinase is about ten times faster than ATP synthesis in mitochondria (∼ 0.7 mmol/s), which is approximately 20 times quicker than ATP generation via glycolysis [226]. Phosphocreatine is the primary energy reserve element in cardiomyocytes. Attenuated phosphocreatine concentration and elevated AMP level activate AMP-activated protein kinase that increases glucose transport and stimulates phosphofructokinase for glycolysis.
Mitochondria-derived ATP is mainly used for cardiac contraction, whereas ATP produced by glycolysis intervenes in activity of kinases and ion pumps and channels (e.g., Ca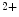 –calmodulin-dependent kinase-2, ATP-sensitive K
–calmodulin-dependent kinase-2, ATP-sensitive K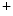 channels, and Na
channels, and Na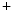 –K
–K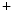 and Ca
and Ca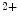 ATPase [SERCA2]) [225].
ATPase [SERCA2]) [225].
–calmodulin-dependent kinase-2, ATP-sensitive K channels, and Na–K and Ca ATPase [SERCA2]) [225].The failing heart has limited energy reserve, i.e., capacity to generate ATP in excess of its normal rate of utilization. The failing heart also has impaired capacity to convert chemical energy into mechanical work. The ratio of phosphocreatine to ATP—an index of energy reserve—wanes.
Although glucose oxidation is more efficient with an ATP production-to-oxygen consumption ratio (ATP/O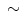 3.1) greater than that of fatty acid (ATP/O
3.1) greater than that of fatty acid (ATP/O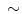 2.8), fatty acid oxidation provides much more ATP (∼129) than glucose (∼ 36) [225].
2.8), fatty acid oxidation provides much more ATP (∼129) than glucose (∼ 36) [225].
3.1) greater than that of fatty acid (ATP/O 2.8), fatty acid oxidation provides much more ATP (∼129) than glucose (∼ 36) [225].The metabolic flexibility (i.e., alternative selection of carbohydrates or lipids according to their circulating concentrations) is altered. Substrate preference shifts from fatty acids to glucose in maladaptive hypertrophy and failing heart, possibly due to dampened expression of fatty acid-handling genes.
In maladaptive hypertrophy and failing heart, three major disturbances in energy handling occur: (1) creatine concentration and creatine kinase activity (Vol. 1, Chap. 4. Cell Structure and Function) lower; (2) glucose uptake by insulin-independent glucose transporter GluT1 and insulin-dependent, dominant GluT4 and glucose metabolism are inadequate, although glucose becomes the main energy provider; and (3) fatty acid oxidation falls [226].
The complex formed by the transcription factor estrogen-related receptor ERRα (or nuclear receptor NR3b1) and peroxisome proliferator-activated receptor PPARγ (or NR1c3) coactivator PGC1α targets a set of genes that encode proteins involved in: (1) energy production, such as those involved in fatty acid and glucose uptake, tricarboxylic acid cycle, and electron transport chain; (2) transfer, such as mitochondrial creatine kinase and adenine nucleotide transporter; and (3) use [226]. In maladaptive hypertrophy and failing heart, cyclin-dependent kinases CDK7 and CDK9 repress PGC1α.
In summary, in advanced stages of ventricular maladaptive remodeling and heart failure, fatty acid oxidation is reduced and more glucose is oxidized. Multiple factors that control both fatty acid uptake and oxidation are inactivated and/or downregulated, such as peroxisome proliferator-activated receptors PPARα (NR1c1), PPARβ (NR1c2), and PPARγ (NR1c3), as well as PPARγ coactivator PGC1α [227].
3.2.2.2 Unsuitable Function of the Electron Transport Chain
Most ATP is synthesized by substrate oxidation in mitochondria that have a high density in cardiomyocytes. In the failing heart, a reduced mitochondrial respiration rate is caused by improper organization of electron transport chain complexes rather than reduced content and activity of complexes of mitochondrial respiratory chains [225].
Mitochondrial uncoupling is detected by a decline in the ratio of mitochondrial ATP production to oxygen consumption. Moreover, failing heart is characterized by decayed expression of mitochondrial genesis factors (PGC1α, nuclear respiratory factors NRF1 and NRF2 (GABP), and mitochondrial transcription factor-A [225].6
Mitochondrial electron transport is a major enzymatic source of oxygen radical generation in cardiomyocytes.7 Altered mitochondria induce oxidative stress in the diseased heart. An elevated level of mitochondrial reactive oxygen species during hypoxia can activate hypoxia-inducible factor HIF1α, thus having beneficial effect [228].
3.2.3 Gene Transcription and Translation
Ventricular hypertrophy is characterized by changes in gene expression that include reactivation of the Anp, Bnp, and βMhc genes of the fetal gene program as well as the suppression of expression of genes that encode proteins implicated in adult heart function [1]. In particular, the αMhc and Serca2a genes expressed at high levels in healthy hearts have a repressed expression during cardiac hypertrophy.
3.2.3.1 Chromatin Remodeling in Cardiac Hypertrophy
Histone acetyltransferase (HAT) and deacetylases (HDAC) determine the expression of cardiac genes (Sect. 1.1.2). Suppression of gene expression also depends on switch/sucrose nonfermentable (Swi/SNF)-related, matrix-associated, actin-dependent regulator of chromatin SMARCa2–SMARCa4-associated HDAC corepressor complexes. The interaction of SMARCa2 with the proximal promoters of the Anp and Bnp genes enhances gene expression, but the recruitment of SMARCa4 to α Mhc promoter causes transcriptional suppression in models of cardiac hypertrophy.
Histone Acetyltransferases
Myocardial stress in the adult heart increases P300 histone acetyltransferase activity as well as histone H3K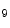 and H4K
and H4K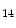 acetylations that provoke activation of genes linked to cardiac hypertrophy [1]. Inhibition of cyclin-dependent kinase CDK9, a mediator of P300–GATA4 complex, attenuates phenylephrine-induced hypertrophy in rat cardiomyocytes. The natural small molecule curcumin is a selective inhibitor of P300 that impedes cardiac hypertrophy-induced acetylation as well as the binding of P300–GATA4 complex at gene promoters.
acetylations that provoke activation of genes linked to cardiac hypertrophy [1]. Inhibition of cyclin-dependent kinase CDK9, a mediator of P300–GATA4 complex, attenuates phenylephrine-induced hypertrophy in rat cardiomyocytes. The natural small molecule curcumin is a selective inhibitor of P300 that impedes cardiac hypertrophy-induced acetylation as well as the binding of P300–GATA4 complex at gene promoters.
and H4K acetylations that provoke activation of genes linked to cardiac hypertrophy [1]. Inhibition of cyclin-dependent kinase CDK9, a mediator of P300–GATA4 complex, attenuates phenylephrine-induced hypertrophy in rat cardiomyocytes. The natural small molecule curcumin is a selective inhibitor of P300 that impedes cardiac hypertrophy-induced acetylation as well as the binding of P300–GATA4 complex at gene promoters.Histone Deacetylases
Histone deacetylases remove acetyl groups from lysine residues in various types of nuclear and cytosolic proteins, especially transcriptional regulators and nucleosomal histones.
Both class-1 (HDAC1–HDAC3 and HDAC8) and class-2 (HDAC4–HDAC7 and HDAC9–HDAC10) are involved in cardiac hypertrophy. In addition, HDACs interact with transcription factors such as myocyte enhancer factor MEF2 and coregulators of expression of genes implicated in cardiac hypertrophy and heart failure.
Different HDAC classes may mediate pro- and antihypertrophic signaling in the stressed myocardium. Class-1 HDAC inhibition can suppress cardiac hypertrophy upon angiotensin-2 infusion and transverse aortic constriction. On the other hand, class-2a HDACs (HDAC4, HDAC5, HDAC7, and HDAC9) repress expression of genes regulated by MEF2 that are involved in cardiac hypertrophy.
In cardiomyocytes, class-1 zinc-dependent histone deacetylases (HDAC1–HDAC3) repress cardiac hypertrophy, as they preclude expression of the gene that encodes dual-specificity phosphatase DuSP5, a nuclear phosphatase that hampers prohypertrophic signaling by dephosphorylation of nuclear ERK1 and ERK2 kinases [229]. Moreover, inhibition of DUSP5 by class-1 HDACs requires activity of the ERK kinase (self-reinforcing mechanism). The HDAC3 subtype regulates ERK1 and ERK2 in cardiomyocytes, whereas HDAC1 and/or HDAC2 controls JNK in these cells.
Phosphatases of the DUSP category8 form the largest group of MAPK phosphatases. The DuSP1 subtype suppresses ERK, JNK, and P38MAPK signaling, thereby impeding cardiac hypertrophy in response to pressure overload [229]. Cardiac-specific overexpression of cytoplasmic DuSP6 promotes cardiac fibrosis and apoptosis in response to pressure overload.
On the other hand, hypertrophic stimuli concomitantly stimulate nuclear ERK1 and ERK2 phosphorylation and repress expression of DUSP5 via class-1 HDACs. Activated cardiac ERK1 and ERK2 cause a concentric cardiac hypertrophy and can be correlated with adverse hypertrophy due to pressure overload in humans [229]. Unlike MAP2K1 overexpression that engenders compensatory hypertrophy, expression of constitutively nuclear ERK2 favors pathological cardiac remodeling in response to pressure overload. Hence, nuclear ERK signaling leads to pathological hypertrophy, whereas phosphorylation of cytosolic substrates by ERK1 and ERK2 ensures cardioprotection.
Both HDAC1 and HDAC2 stimulate cardiac hypertrophy, as they support autophagy [229]. Association of HDAC2 with the Ying Yang YY1 transcription factor promotes expression of BNP in response to a hypertrophic stimulus. In addition, HDAC1 stimulates sodium–calcium exchanger NCX1 expression during cardiac hypertrophy.
3.2.3.2 Reactivation of the Embryonic Program
Maladaptive cardiac hypertrophy is associated with reactivation of a fetal program of cardiac gene expression. The cardiac hypertrophy-associated increase in RNA synthesis is characterized by a reexpression of the NPPA and NPPB genes as well as the fetal myocardial β-myosin heavy chain encoded by the MYH7 gene in cardiomyocyte subsets [230].
β-Myosin heavy chain yields a marker of normal aging as well as pathological cardiac hypertrophy. βMHC+ cells in normal and old as well as hypertrophic hearts are predominantly located in subendocardial clusters within and surrounding fibrosis sites. β-Myosin heavy chain, thereby, is a marker of fibrosis rather than hypertrophy.
3.2.3.3 Elongation Phase of Transcription
The PKB axis promotes protein synthesis and angiogenesis, as it regulates, together with HIF1α, the proangiogenic VEGFa factor produced by cardiomyocytes. In adaptative hypertrophy, angiogenesis matches cardiomyocyte hypertrophy. When angiogenesis is impaired, the heart evolves more rapidly into failure.
The positive (productive) transcription elongation factor-b (pTEFb),9 or cyclin T– cyclin-dependent kinase CDK9,10 and its modulators such as the self-inhibitory RNA kinase and transcriptional repressor hexamethylene bisacetamide-inducible molecule HexIM111 are implicated in cardiac hypertrophy [232]. A reduced expression of HexIM1 is accompanied with an activation of pTEFb. In the catalytically inactive state, PTEFb is complexed with HexIM1 and 7SK small nuclear RNA, among others. In response to various physiological and pathological stimuli such as hypertrophic signals, HexIM1 dissociates from cyclin. Subsequently, PTEFb binds to bromodomain-containing protein BrD4 and, upon phosphorylation of RNA polymerase-2, increases elongation activity and transcription of nascent mRNAs. The affinity of BrD4 to acetylated histones allocates the active pTEFb to specific regions of the genome epigenetically marked as transcriptionally active.
In addition, HexIM1 can directly bind to and modulate transcription factors, such as the estrogen receptor-α (NR3a1), CCAAT/enhancer-binding protein-α, and nuclear factor-κB.
Reactivation of HexIM1 in the adult heart upregulates the protein expression level, in particular that of HIF1α (in addition to MyC, GATA4, and PPARα, among others) and subsequently increasing that of the Vegf gene as well as of genes associated with fatty acid utilization, but decreasing that of genes related to glucose metabolism, without affecting that of stress genes, such as NPPA and NPPB genes [233]. Therefore, HexIM1 induces a cardiac hypertrophy that resembles that linked to exercise rather than that resulting from pressure overload.
3.2.3.4 Transcription Factor NFE2L2
The transcription factor nuclear factor erythroid-derived NFE2-related factor NFE2L2 is protective upon transient activation in response to stress (Vol. 8, Chap. 3. Endothelial Dysfunction). It coordinates the redox balance that conditions microvascular resistance and cardiomyocyte proteins homeostasis [234].
On the other hand, it is detrimental upon prolonged stimulation, engendering proteotoxic cardiac remodeling via reductive stress and toxic protein aggregates as well as maladaptive hypertrophy of cardiomyocytes [235]. Overcompensation of NFE2L2 during cardiac remodeling indeed favors reductive stress and protein ubiquitination and subsequent aggregation, especially that of chaperones and cytoskeletal components, and hence adverse cardiac hypertrophy [235].
A sustained activation of NFE2L2 develops in two phases, initially in response to ROS generation (∼ 3 months), and later, as a consequence of Kelch-like ECH-associated protein KEAP1 dysfunction (6 months) [235].
A NFE2L2 deficiency normalizes the excessive content of glutathione and prevents premature death. In NFE2L2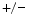 mice (i.e., a single functional copy of the Nfe2l2 gene), the expression of glutathione metabolic and antioxidative enzymes (e.g., NAD(P)H dehydrogenase quinone NQO1, catalase, glucose-6-phosphate dehydrogenase [G6PD],12 and glutathione reductase [GSR]) lowers. Attenuated production of NFE2L2-dependent antioxidant enzymes dampens the reductive stress and reequilibrates the redox milieu [234]. It also restores the production of other redox-sensitive enzymes (e.g., HO1 and
mice (i.e., a single functional copy of the Nfe2l2 gene), the expression of glutathione metabolic and antioxidative enzymes (e.g., NAD(P)H dehydrogenase quinone NQO1, catalase, glucose-6-phosphate dehydrogenase [G6PD],12 and glutathione reductase [GSR]) lowers. Attenuated production of NFE2L2-dependent antioxidant enzymes dampens the reductive stress and reequilibrates the redox milieu [234]. It also restores the production of other redox-sensitive enzymes (e.g., HO1 and 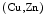 SOD and
SOD and 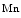 SOD), which falls during reductive stress.
SOD), which falls during reductive stress.
mice (i.e., a single functional copy of the Nfe2l2 gene), the expression of glutathione metabolic and antioxidative enzymes (e.g., NAD(P)H dehydrogenase quinone NQO1, catalase, glucose-6-phosphate dehydrogenase [G6PD],12 and glutathione reductase [GSR]) lowers. Attenuated production of NFE2L2-dependent antioxidant enzymes dampens the reductive stress and reequilibrates the redox milieu [234]. It also restores the production of other redox-sensitive enzymes (e.g., HO1 and SOD and SOD), which falls during reductive stress.3.2.3.5 Hypoxia-Inducible Factor
Heart failure can arise when cardiac angiogenesis does not conform with cardiac hypertrophy [236]. Pressure overload initially promotes cardiac angiogenesis via hypoxia-inducible factor-1. Downregulation of HIF1α causes maladaptive hypertrophy during chronic pressure overload. In maladaptive cardiac hypertrophy, angiogenesis does not reach a sufficient level for the conservation of the cardiac function. Factor P53, the expression of which is upregulated, binds to HIF1α and favors its degradation.
3.2.3.6 Dysregulation in MicroRNA Activity
MicroRNAs are small noncoding RNAs that mediate posttranscriptional gene silencing. The microRNA content in the circulation may reflect the activation state of flowing cells. MicroRNAs circulate in all compartments of blood (plasma, platelets, red blood capsules, and leukocytes; Table 3.4). Circulating microRNAs are stable and resistant to degradation by RNAse. MicroRNAs can then be used as markers of cardiovascular diseases.
Table 3.4
In coronary atherosclerosis, microRNAs have been investigated in serum, plasma, whole blood, peripheral blood mononuclear leukocytes (lymphocytes and monocytes), or agranulocytes, and platelets by different teams of investigators. (Source: [237])
MicroRNAs | Source |
|---|---|
MiR18b, miR22, miR92b, miR126, miR122, miR129-5p, miR-320a, miR423-5p, miR499, miR622, miR654-3p, miR1254 | Plasma, serum |
MiR19b, miR107, miR125b, miR139, miR142-3p, miR142-5p, miR497 | Flowing agranulocytes |
Table 3.5
MicroRNAs in microparticles and their link to cardiovascular diseases. (Source: [239])
microRNA | Platelet microparticle | Monocyte microparticle | Endotheliocyte microparticle | Disease |
|---|---|---|---|---|
Let7d | x | x | CAD | |
MiR17 | x | x | CAD | |
MiR19 | x | x | x | CH, CMP |
MiR20a | x | x | CAD | |
MiR21 | x | x | x | CAD |
MiR27a | x | x | CAD | |
MiR92a1 | x | x | CAD | |
MiR126 | x | x | CAD, DAP | |
MiR130 | x | x | CAD | |
MiR133 | x | x | x | MI, CH |
MiR143 | x | x | CAD | |
MiR146a | x | x | Myocarditis | |
MiR146b | x | Myocarditis | ||
CAD | ||||
MiR155 | x | x | Myocarditis | |
CAD | ||||
MiR199 | x | CAD | ||
MiR221 | x | x | CAD | |
MiR223 | x | x | MI | |
Myocarditis | ||||
MiR423 | x | x | CF |
Circulating microRNAs can reside in vesicles, such as exosomes (50–90 nm), microparticles (size range [100 nm–1.1 μm]), and apoptotic bodies (0.5–2 μm), or bound to lipoproteins (HDLs and LDLs) and proteic complexes (e.g., Argonaute-2 and nucleophosmin-1) [237, 238].
Several microRNAs with high levels in microparticles are involved in cardiovascular diseases [239]. Proinflammatory microparticles (plasma concentration ∼ 5000/μl) are transport carriers for numerous microRNAs, protecting them from degradation [239]. These microRNAs are packaged in producing cells, such as platelets, leukocytes, and endotheliocytes (Table 3.5). They contain cytosol and surface markers of their cells of origin. Once released into the circulation, microparticles bind and fuse with their target cells via receptor–ligand connections. These vectors mediate vascular inflammation and coagulation, due to their specific lipid composition (e.g., phosphatidylserine) as well as to transfer of inflammatory components and microRNAs synthesized in producing cells.
MicroRNA profiles of microparticles differ significantly according to producing cell type and whether the producing cell is stimulated and not, as well as between patients with stable and unstable coronary artery disease [239]. For example, miR21 is one of the highest upregulated miRs in stimulated vs. unstimulated monocytic microparticles. Its level augments under hypoxia in vascular smooth myocytes. Moreover, the miR content of microparticles significantly differs from that of their synthesizing cells.
Type-2 Diabetes
Type-2 diabetes mellitus characterized by chronic elevation of blood glucose levels is one of the major risk factors for cardiovascular disease. Plasma concentrations of miR9, miR15a, miR20b, miR21, miR24, miR28-3p, miR29a, miR29b, miR30d, miR34a, miR124, miR126, miR144, miR146a, miR150, miR191, miR192, miR197, miR223, miR320, miR375, miR486, and miR503 are significantly associated with diabetes mellitus, some being involved in insulin regulation [237].
Cardiac Maladaptive Hypertrophy
During adverse cardiac hypertrophy, several specific microRNAs, especially stress-inducible species, are dysregulated [240]. The expression of miR23a, miR27a, miR24.2, and miR195 is upregulated. In addition, miR21, which impedes apoptosis, is also overexpressed during cardiac hypertrophy. On the other hand, miR133, which represses serum response factor, is underexpressed during cardiac hypertrophy. Overexpression of these microRNAs in cardiomyocytes causes cardiac hypertrophy at least in vitro.
MicroRNA-23a, the production of which is regulated by nuclear factor of activated T cells NFAT3, is a prohypertrophic agent [241]. It targets the ubiquitin–protein ligase tripartite motif-containing protein TRIM63,13 an antihypertrophic protein. MicroRNAs miR23a, miR24, and miR27a belong to the same cluster, but the upregulation of miR24 and miR27a expression occurs later than that of miR23a. In addition, miR23a can stimulate synthesis of early growth response EGR1 and pituitary tumor-transforming gene product PTTG1 (or securin), both involved in cardiac hypertrophy [241].
In addition, circulating levels of miR296-5p, let7e, and 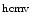 miR-UL112, a human cytomegalovirus-encoded microRNA that targets interferon regulatory factor IRF1, are altered in hypertension [237, 238].
miR-UL112, a human cytomegalovirus-encoded microRNA that targets interferon regulatory factor IRF1, are altered in hypertension [237, 238].
miR-UL112, a human cytomegalovirus-encoded microRNA that targets interferon regulatory factor IRF1, are altered in hypertension [237, 238].Members of the microRNA-34 category (miR34a–miR34c) are upregulated in the heart in response to stress. Therapeutic inhibition of miR34s upregulates [242]: (1) vascular endothelial growth factor that supports coronary angiogenesis; (2) vinculin that preserves cardiac contractility and electrochemical transmission at intercalated discs; (3) protein 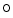 fucosyltranferase POFuT1 that acts in O-linked glycosylation of proteins, hence favoring cell survival; (4) Notch-1 that mediates cardiac regeneration after myocardial infarction; (5) B-cell lymphoma protein BCL6 that protects cardiomyocytes against inflammation; and (6) semaphorin-4B that prevents IL6 production by basophils. Therapeutic inhibition of miR34s attenuates pathological cardiac remodeling [242].
fucosyltranferase POFuT1 that acts in O-linked glycosylation of proteins, hence favoring cell survival; (4) Notch-1 that mediates cardiac regeneration after myocardial infarction; (5) B-cell lymphoma protein BCL6 that protects cardiomyocytes against inflammation; and (6) semaphorin-4B that prevents IL6 production by basophils. Therapeutic inhibition of miR34s attenuates pathological cardiac remodeling [242].
fucosyltranferase POFuT1 that acts in O-linked glycosylation of proteins, hence favoring cell survival; (4) Notch-1 that mediates cardiac regeneration after myocardial infarction; (5) B-cell lymphoma protein BCL6 that protects cardiomyocytes against inflammation; and (6) semaphorin-4B that prevents IL6 production by basophils. Therapeutic inhibition of miR34s attenuates pathological cardiac remodeling [242].The microRNAs miR29b and miR30c are involved in the communication between cardiac fibroblasts and myocytes in myocardial hypertrophy [40]. MicroRNAs act as moderators in the paracrine myocyte–fibroblast crosstalk by mostly regulating the secretion of growth factors and cytokines.
MicroRNA-29b affects matrix protein deposition by fibroblasts as well as causes cardiomyocyte hypertrophy, as it regulates the secretion of messengers, such as IGF1, LIF, and pentraxin-3 [40]. However, a medium derived of miR29b-transfected cardiofibroblasts engenders cardiomyocyte atrophy [40]. On the other hand, miR30c only slightly influences the secretion of matrix constituents, its action being opposite to that of miR29b. A medium derived of miR30c-transfected cardiofibroblasts provokes cardiomyocyte hypertrophy, hence counteracting miR29b effect [40].
As does miR29b, miR133a that is highly expressed in cardiomyocytes targets collagen-1A1 transcripts (COL1A1). Therefore, microRNAs of both cardiac fibroblasts and myocytes synergistically affect COL1A1 mRNA expression, thereby regulating cardiac fibrosis [40].
3.2.4 Signaling Remodeling
Chronic maladaptive hypertrophy of cardiomyocytes can result from valvular dysfunction, persistent hypertension, and myocardial infarction. In particular, left ventricular maladaptive hypertrophy is a response to increased afterload due to systemic arterial hypertension, aortic stenosis, as well as myocardial infarction. Although it tends originally to restore pump function, it leads to heart failure.
The epicardium contains multipotent stem cells and releases paracrine factors for cardiac regeneration and repair. Injury of the adult heart reactivates an epicardial, developmental gene program. Transcription factors of the CCAAT/enhancer-binding protein (C/EBP) category supports neutrophil infiltration after injury and restoration of the cardiac function, especially cardiac remodeling following ischemia [243]. They assist the epicardial production and secretion of retinoic acid, cytokines, and chemokines. They operate with homeodomain-containing transcription factors that are contributors to developmental programs, Hox, Meis, and Grainyhead-like proteins.
In addition to chemical factors and pathways (PP3, CamK, MAPK, PKC, and PKD), mechanical stress triggers cardiac hypertrophy, partly, via activation of auto- and paracrine messengers (e.g., angiotensin and endothelin) that stimulate Gα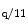 -coupled receptors, including the α1-adrenergic receptors.
-coupled receptors, including the α1-adrenergic receptors.
-coupled receptors, including the α1-adrenergic receptors.Pathological cardiac remodeling is characterized by an increase in cell size, protein synthesis, and sarcomere assembly, as well as reactivation of fetal genes.
3.2.4.1 Calcium Influx
Calcium not only mediates excitation–contraction coupling in cardiomyocytes but also supports the gene transcription that underlies cardiac hypertrophy.
Inositol Trisphosphate Receptors
In addition to ryanodine receptors (RyR), cardiomyocytes also produce inositol trisphosphate receptors (IP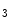 R), but to a lesser extent (approximately 50:1), IP
R), but to a lesser extent (approximately 50:1), IP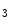 R2 being the main isoform in cardiomyocytes. Nonetheless, Ca
R2 being the main isoform in cardiomyocytes. Nonetheless, Ca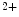 release through IP
release through IP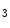 Rs contributes to the inotropic, arrhythmogenic, and hypertrophic effect of Gq-coupled receptor agonists such as endothelin-1.
Rs contributes to the inotropic, arrhythmogenic, and hypertrophic effect of Gq-coupled receptor agonists such as endothelin-1.
R), but to a lesser extent (approximately 50:1), IPR2 being the main isoform in cardiomyocytes. Nonetheless, Ca release through IPRs contributes to the inotropic, arrhythmogenic, and hypertrophic effect of Gq-coupled receptor agonists such as endothelin-1.In cardiac hypertrophy, increased density of IP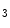 Rs in the junctional sarcoplasmic reticulum augments Ca
Rs in the junctional sarcoplasmic reticulum augments Ca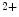 transients [244]. As IP
transients [244]. As IP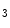 Rs localize close to ryanodine receptors in the junctional sarcoplasmic reticulum, elevated Ca
Rs localize close to ryanodine receptors in the junctional sarcoplasmic reticulum, elevated Ca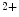 release sensitizes RyRs and increases diastolic and systolic cytosolic Ca
release sensitizes RyRs and increases diastolic and systolic cytosolic Ca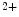 concentration (Ca
concentration (Ca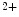 sparks and puffs). These Ca
sparks and puffs). These Ca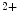 transients can trigger ventricular arrhythmias in cardiac hypertrophy.
transients can trigger ventricular arrhythmias in cardiac hypertrophy.
Rs in the junctional sarcoplasmic reticulum augments Ca transients [244]. As IPRs localize close to ryanodine receptors in the junctional sarcoplasmic reticulum, elevated Ca release sensitizes RyRs and increases diastolic and systolic cytosolic Ca concentration (Ca sparks and puffs). These Ca transients can trigger ventricular arrhythmias in cardiac hypertrophy.However, IP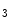 R2 production does not rise in patients with heart failure caused by ischemic dilated cardiomyopathy [244]. On the other hand, atrial fibrillation is also associated with elevated IP
R2 production does not rise in patients with heart failure caused by ischemic dilated cardiomyopathy [244]. On the other hand, atrial fibrillation is also associated with elevated IP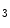 R expression in atriomyocytes in humans.
R expression in atriomyocytes in humans.
R2 production does not rise in patients with heart failure caused by ischemic dilated cardiomyopathy [244]. On the other hand, atrial fibrillation is also associated with elevated IPR expression in atriomyocytes in humans.Calcium Influx Through TRP Channels
The transient receptor potential canonical channels TRPC3 and TRPC6 are produced at low levels in normal conditions. Their expression rises in adverse cardiac hypertrophy. They support hypertrophy signaling triggered by angiotensin-2 or endothelin-1 [245]. A dual inhibition is necessary, as the deletion of the Trpc3 or Trpc6 gene alone does not protect against hypertrophy induced by pressure overload, but combined deletion is protective.
Cardiomyocyte activation by angiotensin-2 or endothelin-1 or by mechanical stress due to sustained hypertension stimulates the Ca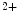 –calmodulin-dependent phosphatase PP3 that dephosphorylates the transcriptional regulator NFAT, thereby provoking nuclear translocation of NFAT and expression of maladaptive hypertrophic genes. The source of triggering Ca
–calmodulin-dependent phosphatase PP3 that dephosphorylates the transcriptional regulator NFAT, thereby provoking nuclear translocation of NFAT and expression of maladaptive hypertrophic genes. The source of triggering Ca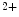 ions is the TRPC3 and TRPC6 channels [245].
ions is the TRPC3 and TRPC6 channels [245].
–calmodulin-dependent phosphatase PP3 that dephosphorylates the transcriptional regulator NFAT, thereby provoking nuclear translocation of NFAT and expression of maladaptive hypertrophic genes. The source of triggering Ca ions is the TRPC3 and TRPC6 channels [245].3.2.4.2 PP3–NFAT Axis
Calcium–calmodulin-dependent protein Ser/Thr phosphatase PP3 dephosphorylates nuclear factor of activated T-cells that can then enter the nucleus and interact with cardiac GATA4 transcription factor involved in cardiac hypertrophy under pathological conditions [265].
The calcineurin (PP3)–nuclear factor of activated T cells (NFAT) signaling operates not only in the nervous system (memory formation or apoptosis) and immunity but also in cardiovascular development (cardiac remodeling as well as cardiomyocyte differentiation and endotheliocyte activation during angiogenesis following inflammation) [246]. The NFAT factor intervenes in physiological and pathological conditions. Many NFAT target genes are involved in cell growth and proliferation as well as in inflammation.14 NFAT signaling depends on the cell type as well as signaling magnitude.
The NFAT family of transcription factors encompasses five members. Among them, four are regulated by the Ca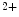 -dependent protein Ser/Thr phosphatase PP3 (NFAT1–NFAT4). The osmotic stress-dependent NFAT5 subtype15 does not depend on calcium ion.
-dependent protein Ser/Thr phosphatase PP3 (NFAT1–NFAT4). The osmotic stress-dependent NFAT5 subtype15 does not depend on calcium ion.
-dependent protein Ser/Thr phosphatase PP3 (NFAT1–NFAT4). The osmotic stress-dependent NFAT5 subtype15 does not depend on calcium ion.In the cytosol, NFAT complexes with the long intergenic noncoding RNA for repressor of NFAT, IQGAP scaffold protein, and importin-β for nucleocytoplasmic transport [246]. Other NFAT partners include BMP2, VEGF, MAGI1, ADAMTS1, thrombin, and histamine.
The NFAT factors are substrates of dual-specificity Tyr phosphorylation-regulated kinase DYRK1a (NFAT-priming kinase), glycogen synthase kinase GSK3β and casein kinase CK1 as well as components of the MAPK module (MAP3K7, MAP3K14, P38MAPK, JNK, and ERK4). Phosphorylation (Ser112) of DSCR1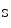 by MAPK leads to the subsequent phosphorylation (Ser108).
by MAPK leads to the subsequent phosphorylation (Ser108).
by MAPK leads to the subsequent phosphorylation (Ser108).Upon dephosphorylation, NFAT undergoes a nuclear translocation. Diverse NFAT family members have redundant functions. The NFAT factors mainly cooperate with other transcription factors, such as AP1, GATA, and MEF2 family members. Otherwise, NFAT has two modes of DNA interaction: (1) binding on NFκB sites (e.g., Vcam1 and Il8) and forming homodimers and (2) binding on a NFAT recognition site as a monomer.
The PP3 phosphatase has many endogenous inhibitors, calcipressins (e.g., calcineurin-binding protein CaBin1, A-kinase anchoring protein AKAP5, and Down syndrome critical region DSCR1 [246]. The DSCR1 regulator prevents PP3 binding to NFAT as well as PP3 activity. Two major DSCR1 isoforms exist: long (DSCR1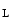 ) and short (DSCR1
) and short (DSCR1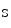 ) variants expressed in the nervous system and in endotheliocytes, respectively, the latter being mainly regulated by the PP3–NFAT pathway and the former by the Notch and HES1-dependent pathways. In endotheliocytes, VEGF-inducible DSCR1 is an effector of a negative feedback loop that hinders NFAT-mediated cell activation and proliferation.
) variants expressed in the nervous system and in endotheliocytes, respectively, the latter being mainly regulated by the PP3–NFAT pathway and the former by the Notch and HES1-dependent pathways. In endotheliocytes, VEGF-inducible DSCR1 is an effector of a negative feedback loop that hinders NFAT-mediated cell activation and proliferation.
< div class='tao-gold-member'>
) and short (DSCR1) variants expressed in the nervous system and in endotheliocytes, respectively, the latter being mainly regulated by the PP3–NFAT pathway and the former by the Notch and HES1-dependent pathways. In endotheliocytes, VEGF-inducible DSCR1 is an effector of a negative feedback loop that hinders NFAT-mediated cell activation and proliferation.Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


