Fig. 3.1
Clinical disease courses in patients with IPF. The typical natural history of IPF is the slow progression of the disease with a median survival period of approximately 3 years from the time of diagnosis (a). In some cases of IPF, abrupt and rapid deteriorations, triggered by unidentifiable causes or obvious causes, occur during the chronic clinical course (b). The deterioration is termed an acute exacerbation of IPF (AE-IPF). AE-IPF can occur even before diagnosis of IPF (c)
3.2 Definition
Diagnostic criteria were first established in 1995 and later revised in 2004 by The Study Group on Diffuse Pulmonary Disorders, Scientific Research/Refractory Disease-Overcoming Research Business, Japan Ministry of Health, Labor and Welfare. According to these criteria, diagnosis requires the following: (1) progressive dyspnea over 1 month or less, (2) new pulmonary infiltrates seen on a high-resolution computed tomography (HRCT) scan with evidence of underlying usual interstitial pneumonia (UIP), (3) worsening hypoxemia (i.e., a fall in PaO2 of >10 mmHg), and (4) the absence of an underlying cause such as pulmonary infection, pneumothorax, malignancy, pulmonary embolism, and cardiac failure. A study group in the United States reported a similar definition in 2007 [10] (Table 3.1), which includes an item to exclude pulmonary infection by endotracheal aspirate or bronchoalveolar lavage (BAL) to this criterion. Although the presence of overt infection is considered to be an exclusion criterion, occult infection could be a trigger factor for AE-IPF. In fact, bacterial and viral pneumonias are often seen in patients with AE-IPF and may be the trigger factor. Therefore, although it is important to exclude simple pulmonary infection from AE-IPF, the existence of infection should not preclude the diagnosis of an AE [12].
Table 3.1
Diagnosis of acute exacerbation
Diagnostic criteria |
|---|
Previous or concurrent diagnosis of idiopathic pulmonary fibrosisa |
Unexplained worsening or development of dyspnea within 30 days |
High-resolution computed tomography with new bilateral ground-glass abnormality and/or consolidation superimposed on a background reticular or honeycomb pattern consistent with usual interstitial pneumonia patternb |
No evidence of pulmonary infection by endotracheal aspirate or bronchoalveolar lavagec |
Exclusion of alternative causes, including the following: |
Left heart failure |
Pulmonary embolism |
Identifiable cause of acute lung injuryd |
In AE-IPF without a prior diagnosis of IPF, differentiation from acute interstitial pneumonia (AIP) represents an important diagnostic challenge. However, if the typical honeycomb findings of IPF are observed in the lungs by HRCT, an AE-IPF is diagnosable.
3.3 Pathophysiology
AE-IPF is considered to be a sudden acceleration of the disease or acute lung injury by an unknown cause that is superimposed on the diseased lung. Pathologically, AE-IPF is characterized by an acute-onset diffuse alveolar damage (DAD) that develops close to chronic UIP lesion. Given that DAD is also a common pathological feature of acute respiratory distress syndrome (ARDS) and AIP, it is possible to regard AE-IPF as ARDS due to IPF. In IPF, the lung tissue without fibrosis is primed by inflammatory cytokines produced by chronic inflammation, and it seems to be susceptible to develop DAD through some unknown trigger. Therefore, it is easy to understand AE-IPF as complication of ARDS-like DAD in IPF rather than as deterioration of IPF itself. This is supported by the fact that AE occurs not only in IPF (UIP) but also in other chronic interstitial pneumonias such as nonspecific interstitial pneumonia [13, 14].
3.4 Trigger Factors
Although the triggers for AE-IPF are often unclear, surgical operation, corticosteroid dose reduction, and viral infection can present as obvious triggers [11]. Diagnostic procedures such as BAL and video-assisted thoracoscopic surgery (VATS) are also potential triggers [15]. Gastroesophageal reflux disease (GERD) is another potential cause of AE-IPF [10].
According to the diagnostic criteria, pulmonary infection should be excluded; however, in practice, viral and bacterial infections are often obvious triggers [16, 17]. Simon-Blancal et al. [18] demonstrated that AE-IPF was more frequent in winter and spring than during summer and fall, suggesting that unidentified infections might be an important trigger. Despite this, a study using standard PCR analysis of BAL fluid demonstrated that common respiratory viruses were detected in only four of the 43 patients with AE-IPF, with no evidence of viral infection in most cases [19]. However, limitations with this finding mean that viruses cannot be definitively excluded as the cause of AE-IPF [20].
Some medications have been reported to be triggers for AE-IPF. Implicated drugs include biologic (anakinra, etanercept, and infliximab), nonbiologic (ambrisentan), immunomodulatory (interferon alpha/beta, everolimus, and leflunomide), and anticancer agents [21–23]. In fact, patients with lung fibrosis are prone to drug-induced lung injury. However, because drug-induced DAD-type lung injury can develop in patients without lung fibrosis, it is debatable whether such cases should be regarded as drug-induced AE-IPF or simply as comorbid drug-induced lung injury.
Pulmonary resection in patients with lung cancer and interstitial lung disease can provoke AE-IPF at higher rates and with higher mortality. A systematic review showed that the incidence of postoperative AE-IPF ranged from 0 to 20.8 %, with mortality ranging from 37.5 to 100 % [24]. A large-scale multi-institutional cohort study reported that AE-IPF occurred in 164 of 1763 (9.3 %) patients with non-small cell lung cancer who underwent pulmonary resection and that it was the leading cause of 30-day mortality (71.7 %), with an overall mortality rate of 43.9 % when it developed [25]. Surgical procedures show the strongest association with AE-IPF; for example, using wedge resection as a reference, lobectomy or segmentectomy has an odds ratio of 3.83, and bi-lobectomy or pneumonectomy has an odds ratio of 5.70 (P < 0.001). In high-risk patients, surgical procedures associated with a higher risk of AE-IPF should be chosen cautiously. No benefit was found for perioperative steroids and sivelestat prophylaxis in that study.
Suzuki H et al. [26] analyzed the HRCT findings of patients with IPF to identify radiological characteristics of IPF susceptible to acute exacerbation after surgery for lung cancer. They demonstrated that the degree of fibrosis on preoperative HRCT was significantly higher in the exacerbation group (P < 0.003).
3.5 Epidemiology
The incidence of AE-IPF varies greatly between studies (8.5–60 %), and the precise determination is made difficult by the use of retrospective analyses of selected cases. The incidence also changes depending on whether pulmonary infection is completely excluded. AE-IPF is also increasingly common because of the spreading recognition that it is a common clinical feature of IPF. Recent data from randomized controlled trials have provided incidences that are more conservative, with rates of 14 % over 9 months and 4.8, 5.4, and 9.6 % over 1 year in the control groups [27–29]. Using retrospective data from a large observational cohort of 461 patients, 1- and 3-year incidences are suggested to be 14 % and 21 %, respectively [11]. A population-based analysis demonstrated the rate of AE was 0.13 cases per person year [30].
There are no significant differences in disease duration, pulmonary function, age, gender, or smoking history between patients with and without AE [9, 31]. By multivariate analysis, Song et al. [11] demonstrated that low forced vital capacity (FVC) levels and the absence of a history of smoking were risk factors for AE-IPF; however, the role of smoking in AE-IPF is controversial [32, 33]. Pulmonary hypertension may also be a risk factor for AE-IPF [34].
The mortality rates reported in small case series have been very poor, and they are as high as 85 % [7, 35–39]. In a summary of 16 studies, Collard et al. [10] reported an overall mortality rate of 70 %, while a systematic review reported 1- and 3-month mortality rates of 60 % and 67 %, respectively [40]. Kishaba et al. [41] reported that extensive disease on chest HRCT, including traction bronchiectasis, honeycombing, ground-glass opacity, and consolidation, was associated with particularly poor mortality in AE-IPF. Indeed, the 3-month mortality was 80.6 % among patients with extensive HRCT-related disease findings, which compared negatively with the mortality of 54.5 % in patients with limited disease (P = 0.007).
3.6 Histopathology
The histopathology of AE is characterized by an underlying fibrotic interstitial pneumonia with superimposed DAD [9, 10]. The latter usually appears in a relatively normal area without prior honeycombing, and it is histologically the same as DAD that occurs without a background of UIP such as that observed in AIP and ARDS. Kang et al. [42] investigated the pathological differences depending on the underlying risk, determining the degree of α-smooth muscle actin-positive or collagen type I-positive alveolar interstitial myofibroblasts in the proliferative phase of DAD by immunohistochemical staining. Only two of seven patients with septic ARDS showed interstitial myofibroblast proliferation as opposed to 15 of 16 patients with drug-induced ARDS. Only three patients had AIP, but all patients showed myofibroblast proliferation. These findings indicated that DAD can be divided into two subphenotypes: less fibrogenic and more fibrogenic. The former is seen in patients with septic ARDS and a high incidence of multi-organ dysfunction syndrome (MODS), while the latter is seen in those with drug-induced ARDS, AIP, and AE-IPF with a low incidence of MODS.
3.7 Radiological Assessment
The most common radiological finding in patients with AE-IPF is the presence of new bilateral ground-glass opacities or of consolidation superimposed on the underlying UIP (i.e., subpleural reticular and honeycombing densities) [43]. Acute exacerbations of either IPF or other chronic interstitial lung diseases can closely resemble ARDS in both clinical presentation and chest radiographic abnormalities. Similar to ARDS, pathological findings are dominated by DAD, although the prognosis is substantially worse. The diagnosis of AE-IPF is suggested by a careful review of previous chest radiographic images, by the discovery of subpleural reticular changes intermixed with alveolar opacities on a chest CT scan obtained shortly after the onset of ARDS, or by surgical lung biopsy.
Akira et al. [44] classified the new onset parenchymal abnormalities into three patterns: peripheral, multifocal, and diffuse (Fig. 3.2). By multivariate analysis, the strongest correlations were observed between CT patterns (combined diffuse and multifocal versus peripheral) and survival (odds ratio, 4.629; P = 0.001). Diffuse and multifocal ground-glass patterns appear to predict a worse survival in patients with AE-IPF compared with those with peripheral patterns. In contrast to these findings, Silva et al. [45] were unable to show any relationship between radiographic pattern and survival. Data that are more recent suggest that the extent of lesion on HRCT is a more important determinant of the outcome than the distribution of lesion [46]. Using the HRCT score, which was calculated based on normal attenuation areas and extent of abnormalities (i.e., areas of ground-glass attenuation and/or consolidation with or without traction bronchiectasis or bronchiolectasis and areas of honeycombing), survival among patients was worse with an HRCT score of ≥245 than those with a lower score (log-rank test, P < 0.0001).
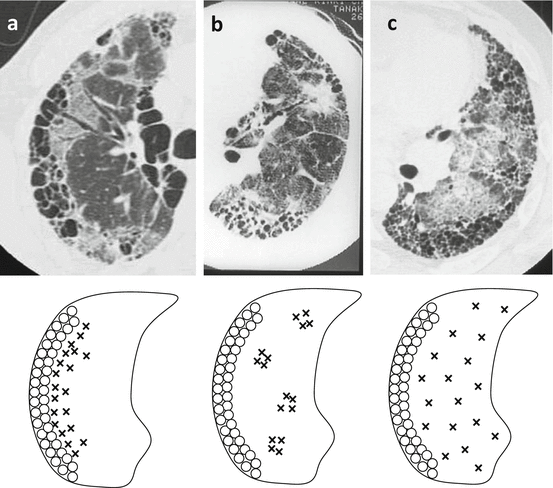
Fig. 3.2
High-resolution computed tomography (HRCT) patterns of acute exacerbation of IPF: (a) peripheral pattern, (b) multifocal pattern, (c) diffuse pattern (Adapted from Ref. [44])
3.8 Laboratory Tests and Biomarkers
When the acute exacerbation occurs, cough and dyspnea become acutely worse within one month. Blood test typically shows increases in the white cell count and both C-reactive protein and lactate dehydrogenase levels. In addition, markers of fibrosis are increased, including surfactant protein-A (SP-A), surfactant protein-D (SP-D), and sialylated carbohydrate antigen (KL-6). Measurement of serum KL-6 level is very useful when assessing disease activity and prognosis in IPF, and it is a useful predictive marker for AE-IPF. Ohshimo et al. [47] reported that baseline serum KL-6 levels were significantly higher in patients who later developed AE-IPF than in those with stable IPF (P < 0.0001). At a KL-6 cutoff level of 1300 U/mL, the sensitivity, specificity, accuracy, and likelihood ratio for predicting AE-IPF were 92 %, 61 %, 66 %, and 2.36, respectively. In a Kaplan-Meier analysis, they reported that patients with baseline serum KL-6 levels of ≥1300 U/mL experienced earlier onset of AE-IPF (P = 0.002). Thus, it is suggested that baseline serum KL-6 (both continuous and at a cutoff level of ≥1300 U/mL) is a sensitive independent predictive factor for the onset of AE-IPF. However, it is essential that care be taken while prescribing steroids or immunosuppressive therapy in IPF because of the risk of opportunistic infections by pneumocystis pneumonia or cytomegalovirus. In patients with pneumocystis pneumonia, although serum KL-6 rises in a similar manner to AE-IPF, blood β-d-glucan levels rise concurrently.
Pneumonia represents the most problematic differential in the diagnosis of AE-IPF. According to the current diagnostic criteria, pulmonary infection must be ruled out by endotracheal aspiration or BAL. However, performing bronchoscopy in patients with AE-IPF is associated with a high risk of worsening the respiratory condition, with BAL itself recognized to be a trigger factor for AE-IPF. In practice, it is often difficult to differentiate between AE-IPF and bacterial pneumonia, and a surrogate marker has, therefore, been sought to exclude infectious pathology. To this end, serum procalcitonin (PCT) is useful while attempting to differentiate between typical bacterial and nonbacterial causes of inflammation. Nagata et al. [48] reported that serum PCT levels in AE-IPF were significantly lower than those in bacterial pneumonia with IP (0.62 ± 1.30 vs. 8.31 ± 14.83 ng/mL; P < 0.05). Thus, serum PCT is a useful surrogate marker for discriminating between AE-IPF and concurrent bacterial pneumonia with IPF.
In addition, circulating fibrocytes have been reported to be a good biomarker of fibrosis. Fibrocytes are circulating bone marrow-derived spindle-shaped cells, produce extracellular matrix components, and may play an important role in wound repair and tissue fibrosis. Fibrocytes were defined as cells positive for CD45 and collagen-1 by flow cytometry. Circulating fibrocytes defined as CD45-positive and collagen-1-positive cells by flow cytometry are increased threefold in patients with stable IPF compared with healthy controls [49]. During AE-IPF, fibrocyte counts have been shown to further increase to an average of 15 % of peripheral blood leukocytes. The increased fibrocyte counts then tend to decrease to pre-exacerbation levels in patients who recover. Circulating fibrocyte counts can also indicate prognosis; patients with IPF and fibrocytes of >5 % had a poor prognosis when compared with those with fibrocytes of <5 % (mean survival time, 7.5 months vs. 27 months; P = 0.0001).
3.9 Pharmacological Treatments
To date, no evidence-based effective therapy has been established for AE-IPF. Indeed, international consensus guidelines produced by the ATS, ERS, Japanese Respiratory Society, and Latin American Thoracic Association only recommend management supportive care and corticosteroid use [50].
Empirical treatment with high-dose corticosteroid therapy is generally used in AE-IPF without any clear evidence that they are effective. No randomized controlled trials have been conducted using corticosteroids in AE-IPF. Methylprednisolone pulse therapy [1 g intravenously (IV)] is usually given on days 1–3 with a maintenance prednisolone dose equivalent to approximately 40 mg daily [5]. The 3-day pulse therapy is repeated weekly on 1–4 occasions until the patient’s condition stabilizes.
Steroid pulse therapy came to be used for respiratory diseases based on its biological plausibility and experience, with a similar method used for renal and collagen diseases [51]. In Japan, this therapy has been used for IPF and AE-IPF since 1978. It is also usually used in the treatment of other lung diseases presenting with DAD, such as ARDS and AIP. Although the rapidly progressive clinical condition is often temporarily stabilized by the pulse therapy, thereby improving oxygenation, there is no evidence that it improves mortality. Indeed, the mortality rate remains high, despite therapy. In recent years, a low-dose steroid therapy has been employed (methylprednisolone, 1 mg/kg/day) in reference to trials in the treatment of ARDS [52], but the benefits remain unclear. Given that the pathological evidence of DAD is an extremely poor prognostic factor, it seems that the effect of steroids may be insufficient. Nevertheless, surgical lung biopsies reveal that some cases of AE-IPF have organizing pneumonia rather than DAD [53], in which case glucocorticoid therapy might be effective.
Immunosuppressants, such as cyclosporine A (CsA), cyclophosphamide, or tacrolimus, are used together when the reaction of steroid alone is poor. These drugs can also be used in combination with a steroid from the beginning. Several investigators reported better survival in patients treated with this type of combination therapy [54–56]. Although these studies have mostly been retrospective, have included small samples, and have used various definitions of AE-IPF, they suggest that the use of immunosuppressants in combination with corticosteroids is more effective than corticosteroid monotherapy. Sakamoto et al. [54] reported that the mean survival period after the first onset of AE-IPF was 285 days in a CsA-treated group and 60 days in a non-CsA-treated group. Thus, prognosis was significantly better in the CsA-treated group. In this study, a low dosage of CsA (100–150 mg/day) was started at the same time as the pulse therapy. Morawiec et al. [55] reported their experience with cyclophosphamide pulse therapy for AE-IPF in a small case series. Patients with AE-IPF were treated with a methylprednisolone pulse (1,000 mg) on days 1–3, before the escalating regimen of cyclophosphamide was started on day 4 with an initial intravenous dose of 500 mg. The dose of cyclophosphamide was then increased by 200 mg every 2 weeks up to the maximum dose of 1,500 mg.
The majority of patients with AE-IPF receive empiric broad-spectrum antibiotics against respiratory pathogens, even if there is no obvious infection. This is based on the following clinical rationale: (1) an underlying infection can be easily missed on microbiological testing, (2) the mortality rate is very high, (3) many patients present with fever and flu-like symptoms and have elevated blood neutrophil counts and CRP levels, and (4) antimicrobial therapy has a low risk of complications.
Because a coagulation disorder subsequent to vascular injury could be important for the pathogenesis of AE-IPF, anticoagulant therapy may be effective. A small prospective clinical trial of anticoagulation with warfarin and low molecular weight heparin in patients with IPF reportedly improved survival in the group receiving anticoagulation mostly by reducing the mortality associated with AE-IPF [57]. In that study, the mortality associated with AE-IPF was significantly reduced in the anticoagulant group when compared with the non-anticoagulant group (18 % vs. 71 %, respectively; P = 0.008). Conversely, a randomized, double-blind, placebo-controlled study of warfarin as a treatment for IPF had to be terminated early because of higher mortality in the warfarin arm and a low likelihood of benefit [58]. Therefore, at present, anticoagulant therapy in patients with IPF is discouraged.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


