Fig. 2.1
Distribution of left ventricular ejection fraction measured in women and men enrolled in the EuroHeart Failure survey (From Cleland et al. [4])
The incidence of HF and its trends are highly variable. The incidence raw (not adjusted for age) in the general population ranges from 1 to 5 cases per 1,000 person-years (28–34), while the data from the largest population-based studies report an incidence ranging from 1 to 2 per 1,000 cases per year. The wide variability in the data of incidence is largely due to the use of diagnostic criteria is not unique and only partially defined. In addition, the incidence data could be made further inaccurate by several factors such as the low percentage of patients autopsied, the economic interest in excluding HF as a discharge diagnosis tab nosographic and the difficulty of framing this syndrome as a primary diagnosis or secondary. One datum definitely ascertained is represented by the exponential increase in the incidence of HF with advancing age (Fig. 2.2). With regard to changes in the incidence of HF in time, the Framingham Heart Study showed only a slight decline in incidence during the last three decades, although it must be stated that this study was prior to the use of ACE inhibitors or thrombolytics.
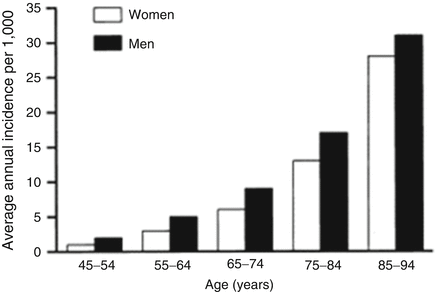
Fig. 2.2
The exponential increase in the incidence of HF with advancing age
The prevalence of HF is progressively increasing due to the aging of the general population. It is estimated that today the 9.1 % of individuals older than 80 years present a picture of HF and that in the future this percentage is set to grow further. In the United States, it was estimated that in 1997 people aged over 65 years were 33 million (of which about 7.9 million aged greater than or equal to 80 years) and that, by the year 2030, this number will increase to approximately 70 million (of which 18 million aged greater than or equal to 80 years). It may therefore be expected, even with conservative estimates, that, by that time, the number of elderly patients with HF will double, reaching a value of 3.6 million. The prevalence of HF varies from 3 to 20 individuals per 1,000 people, with higher figures for individuals over the age of 65 years.
Before the modern era of treatment, 60–70 % of patients died within 5 years of diagnosis and 13.5 % died between admission and 12 weeks follow-up (Fig. 2.3). And there was frequent and recurrent admission to hospital: within 12 weeks of discharge, 24 % of patients had been readmitted (Fig. 2.4). Effective treatment has improved both of these outcomes, with a relative reduction in hospitalization in recent years of 30–50 % and smaller but significant decreases in mortality [4–7].
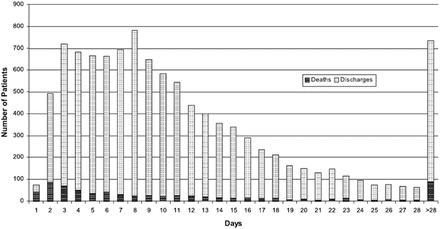
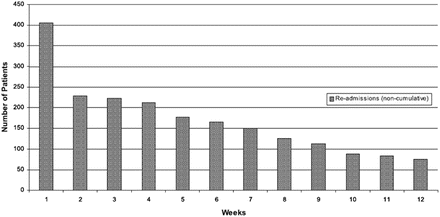
Fig. 2.3
Deaths on index admission and discharges from the time of admission (From Cleland et al. [4])
Fig. 2.4
First admission over 12 weeks for any reason from the time of index admission discharge (From Cleland et al. [4])
Aetiology of Heart Failure
The most frequent causes of HF are represented by coronary artery disease (CAD is the cause of approximately two-thirds of cases of systolic HF), cardiomyopathy and hypertension (HBP), while valvular heart disease, congenital heart disease are more rare (Fig. 2.5). Other causes of systolic HF can be: previous viral infection (recognized or unrecognized), alcohol abuse, hemotherapy (e.g. doxorubicin or trastuzumab), and ‘idiopathic’ dilated cardiomyopathy (although the cause is thought to be unknown, some of these cases may have a genetic basis) [8]. According to data from the Framingham study HBP, associated or not with ischaemic heart disease, is the most common cause of HF in the United States. By contrast in Europe, as reported from studies conducted in England and Sweden, the predominant cause of HF is represented by chronic ischaemic heart disease, HBP or cardiomyopathy represent the etiology of HF in percentages lower than 10 %. The data relating to SEOSI, observational epidemiological study conducted in Italy on HF in a population of nearly 4,000 patients referred to hospital centers specialize, identified in the etiology of ischaemic heart disease more frequent with a percentage of 42 % of patients while a role less obvious is found for HBP (20 %), dilated cardiomyopathy (15.3 %) and valvular heart disease (14 %), respectively [9]. Among the plausible reasons for the discrepancies classificative in terms of etiology of HF are certainly numbered the mode of interpretation of the results of epidemiological studies. In particular the role of arterial hypertension is certainly prevalent in all those conditions as the Framingham study in which the development of HF is related to the finding of HBP in each phase of the natural history regardless of the fact that the same has acted as a risk factor for the development of ischaemic heart disease.
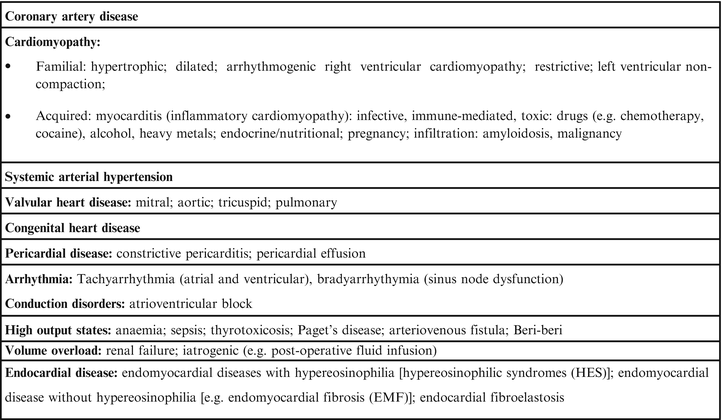
Fig. 2.5
Causes of HF
In contrast, the role of the same HBP is greatly reduced from those studies (mainly in Europe) in which the development of HF is attributed to the ultimate cause that is responsible for it (e.g., chronic ischaemic heart disease, myocardial infarction [MI] or cardiomyopathy) regardless of the presence anamnestic or clinic HBP.
Pathophysiology of Heart Failure
HF is a complex syndrome with a multifactorial genesis characterized by an inability of the heart to adapt to changes in the metabolic needs of the tissues and supported by hemodynamic changes and different neurohormonal systems, in which the symptoms related to reduced functional capacity and the water retention dominate the clinical picture accompanied with reduced survival. HF can be achieved with alterations in pump function or systolic or diastolic function or filling or, as more often happens, both resulting mainly depression of intrinsic ventricular contractility or changes in mode of contraction. Through the therapeutic restoration of intrinsic contractility of the myocardium can get the simultaneous improvement of systolic function and diastolic function.
Besides the reduction of the intrinsic contractility, a further primary cause of depression of ventricular function can also be the asinergia that makes uneven and asymmetric, and therefore asynchronous, the contraction of the ventricular myocardium for the presence of areas which are contracted little or nothing (zones of hypokinesia and akinesia) or which are contracted with excessive delay (asynchronous areas). The asynchronous contraction of the myocardium, mostly due to ischemic infarction or ventricular arrhythmias, depresses the pump function of the ventricle.
The appearance of alterations of myocardial function affects the development of a series of adaptation mechanisms functional, structural and neurohormonal which are initially able to compensate for the impaired myocardial, but that in a second time can represent elements responsible for a further progression of the disease.
In the initial phase of HF, all conditions characterized by an impaired intrinsic contractility (or inotropism), by distensibility (compliance), by the synergy of contraction of the ventricular walls, by an excessive hemodynamic load or by the association of some of these conditions, induce the heart to resort to various compensatory mechanisms of adaptation, immediate or delayed, aimed to preserve its pump function.
If the overload systolic or diastolic are not removed, the phase of functional insufficiency follows a second phase of re-structural adaptation, characterized by a stimulation of the synthesis of myocytes, resulting in hypertrophy (and according to some authors, also hyperplasia) of the muscle cells and hyperplasia of interstitial component, mainly fibroblasts and matrix collagen. The wall stress also causes a stimulus to gene expression involving oncogenes, myocardial protein (ANP, BNP, angiotensin II). The combination of these processes conditions the development of a parietal hypertrophy. In the terminal stages of HF the maladaptive changes occurring in surviving myocytes and extracellular matrix after myocardial injury (e.g. MI) lead to pathological ‘remodelling’ of the ventricle with dilatation and impaired contractility, one measure of which is a reduced ejection fraction (EF), that it is a sign of LV systolic dysfunction [10, 11].
What characterizes untreated systolic dysfunction is progressive worsening of these changes over time, with increasing enlargement of the LV and decline in EF. Two mechanisms that underlie these events: the occurrence of further events leading to additional myocyte death (e.g. recurrent MI) and the systemic responses induced by the decline in systolic function, particularly neurohumoral activation. Two key neurohumoral systems activated in HF are the renin–angiotensin–aldosterone system and sympathetic nervous system. Initially this neuro-hormonal mechanisms have a compensatory function, aimed at maintaining an adequate perfusion to vital organs, but in the long term influence a number of physiologic abnormalities counterproductive as the retention of sodium and water, peripheral vasoconstriction and degenerative processes of myocardial muscle. In addition to causing further myocardial injury, these systemic responses have detrimental effects on the blood vessels, kidneys, muscles, bone marrow, lungs, and liver, and create a pathophysiological “vicious cycle” (Fig. 2.6), accounting for many of the clinical features of the HF syndrome, including myocardial electrical instability. Interruption of these two key processes is the basis of much of the effective treatment of HF [10, 11].
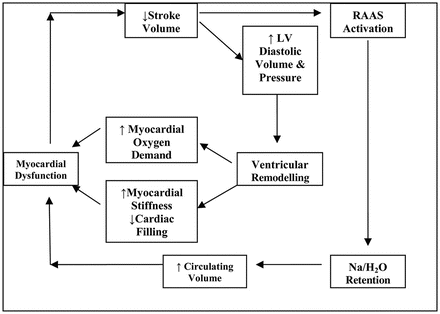
Fig. 2.6
The “vicious cycle” of the HF
In this contest it is clear that the renin-angiotensin system (RAS) plays a central role in the pathophysiology of HF. Therefore to know the mechanisms that underlie this system is an important element to understand and choose the best therapeutic strategy for HF.
The Renin-Angiotensin System
The RAS contributes in the contest of the HF to the increase of the peripheral vascular tone and hydrosaline retention concomitantly with the activation of the sympathetic nervous system. The reduction in cardiac output that characterizes HF causes an increase in plasma renin activity, levels of angiotensin II and aldosterone, which contribute to the development of the adverse effects that characterize HF. In Fig. 2.7 are depicted the different routes of production of angiotensin II which results from the activation of the system over that in circulating level also by an activation of the same at the tissue level, with local production of angiotensin II capable of performing a action vasoactive and trophic. The extent of activation of plasma ACE may reflect incompletely and partially the corresponding tissue activity in particular in patients with HF. Infact in this patient, from the very early stages, it could be observed a predominant local activation of the RAS with production of angiotensin II, even for alternative ways of production and non-employees from the ACE (es. chimasi) that seem particularly important at the tissue level where they could be responsible for the production of angiotensin II by up to 90 %. The RAS is, as can be imagined, also activated in the heart, where it has been hypothesized to contribute to ventricular remodeling phenomena described in the previous paragraph. Indeed angiotensin II is able to stimulate the growth of cardiomyocytes, in turn facilitated by the release of norepinephrine induced by angiotensin II at the level of the sympathetic nerve endings. The biological actions of angiotensin II are realized through the interaction with 4 subtypes of receptors called AT1-AT4, but at present most of the effects of angiotensin II appear mediated by the AT1 receptor, while for the AT2 receptor have been hypothesized antiproliferative and vasodilators effects. The blockade of the AT1 receptor inhibits the action of angiotensin II at the receptor level, and allows a more efficient blockade of angiotensin II. In particular, one of the dominant effects of angiotensin II is represented by the stimulus to the production of aldosterone which has assumed great importance in patients with HF because of its ability to stimulate the reabsorption of sodium, but especially to induce the development of myocardial fibrosis with consequent the progression of myocardial structural alterations described in the previous paragraph. These changes are directly related to the progression of HF in hemodynamic level.
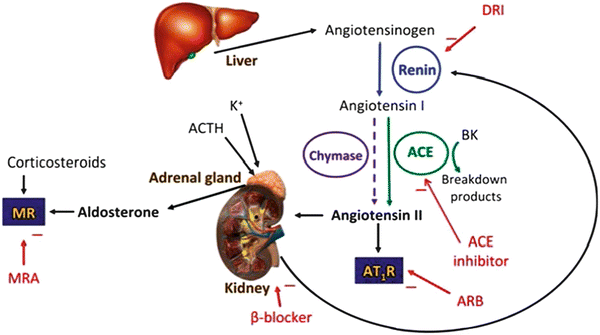
Fig. 2.7
The classical renin–angiotensin–system. DRI direct renin inhibitor, ARB angiotensin receptor blocker, MRA mineralocorticoid receptor antagonist, K + potassium ion, ACE angiotensin converting enzyme, ACTH adrenocorticotropic hormone (corticotropin), BK bradykinin, AT 1 R angiotensin II type 1 receptor, MR mineralcorticoid receptor (From McMurray [12])
Focus on Blockade of the Renin–Angiotensin System
The objectives of the treatment of HF are varied and represented by the reduction of the symptoms, the prevention of the progression of the disease, by improving the quality of life, reduction in the frequency of hospitalization and especially by the prolongation of survival. In particular, the availability of drugs able to effectively interfere with the neurohumoral activation has allowed antagonize or modulate some of these systems to localization cardiac and extracardiac and responsible for the onset, the clinical expression and progression of the disease. In this context, ACE inhibitors represent the class of drugs most widely used among those used in the treatment of HF.
The clinical efficacy of ACE inhibitors follows to the unique mechanism of action that is articulated in an inhibition of the production of angiotensin II (potent vasoconstrictor and growth factor) which is associated with an inhibition of the degradation of the vasodilator bradykinin features of property resulting from the release of nitric oxide and prostacyclin. ACE inhibitors also reduce the activity of the sympathetic nervous system by inhibiting the action of angiotensin II which is capable of promoting the release of norepinephrine and inhibit the resorption (re-uptake). In addition, drugs of this class cause an increase in the density of the ß-adrenergic receptors (through mechanisms of up-regulation) and improve the heart rate variability, the response of the baroreceptor and autonomic function (including the vagal tone).
ACE inhibitors also exhibit antiproliferative effects (reduction of vascular and cardiac hypertrophy and extracellular matrix proliferation) and reduce ventricular remodelling after myocardial infarction [13, 14]. In the hypertrophied heart reduce cardiac hypertrophy and improve diastolic function.
Moreover ACE inhibitors decrease renal vascular resistances and increase renal blood flow and promote Na+ and water excretion by the relatively greater effect in dilating postglomerular efferent than afferent arterioles, leading to a reduction in glomerular capillary hydrostatic pressure and glomerular filtration rate (GFR) [15]. So prevent progression of microalbuminuria to overt proteinuria [16], attenuate the progression of renal insufficiency in patients with a variety of non-diabetic nephropathies [17] and prevent or delay the progression of nephropathy in patients with insulindependent diabetes mellitus [18, 19].
In most patients ACE inhibitors are well tolerated, however, several adverse reactions may occur. They can also appear at any time during treatment, even in patients already chronically treated with ACE inhibitors. The most common adverse reaction associated with their use in the elderly population is orthostatic hypotension (prevalence, ~ 50 %), especially during the first few days of treatment or after a dose increase. Dry cough appears in 5–10 % of patients, this is the most common adverse reaction associated with increased concentration of kinins, and is not dose-dependent. If the cough persists and interferes with quality of life, therapy with ACE inhibitors may be suspended and replaced by the administration of angiotensin II receptor blockers. Hyperkalemia due to a decrease in aldosterone secretion is rarely found in patients with normal renal function but it is relatively common in those with congestive HF and in the elderly. This side effect is also more frequent in patients with renal impairment, diabetes, receiving either K+ or potassium K+-sparing diuretics, heparin or Non-Steroidal Anti-Inflammatory Drugs (NSAIDs). Angioedema is a rare but potentially life-threatening and appears related to an accumulation of bradykinin. Symptoms range from mild gastrointestinal disturbances to severe dyspnea and death. Finally ACE inhibitors, taken during the second or third trimester of pregnancy, may present some teratogenic effects.
Trials That Support the Use of Angiotensing-Converting Enzyme Inhibitors
The evidence supporting the use of ACE inhibitors in patients with HF is based on the results of wide prospective clinical studies (Fig. 2.8). This trials have demonstrated and repeatedly confirmed that ACE inhibitors are effective in reducing morbidity and mortality and are also able to improve the quality of life in patients with asymptomatic LV dysfunction or suffering from a overt congestive HF resulting from a reduced systolic function of the LV or when it is a result of a MI.
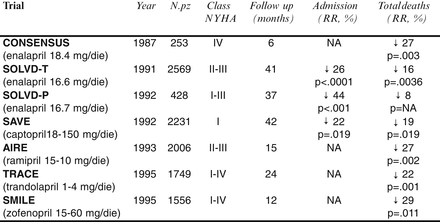
Fig. 2.8
Main trials on ACE inhibitors. NA not available
Two key randomized controlled trials [Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS) [20] and Studies of Left Ventricular Dysfunction (SOLVD)-Treatment] [21] assigned about 2,800 patients with mild to severely symptomatic HF to placebo or enalapril. This trials show how the addition of enalapril to conventional therapy in patients with severe congestive HF can reduce mortality and improve symptoms. The beneficial effect on mortality is due to a reduction in death from the progression of HF (Fig. 2.9).
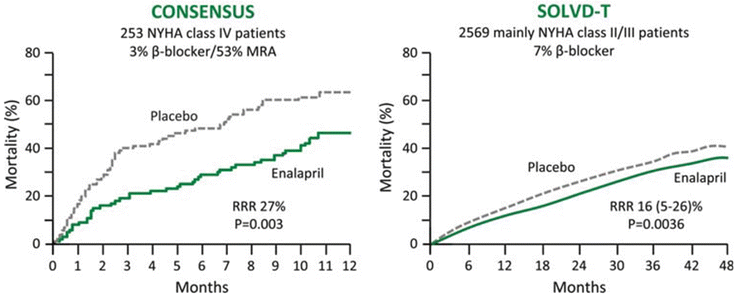
In particular the CONSENSUS evaluate the influence of the angiotensin-converting-enzyme inhibitor enalapril (2.5–40 mg per day) on the prognosis of severe congestive HF (New York Heart Association [NYHA] functional class IV). The trial randomizes 253 patients in a double-blind study to receive either placebo (n = 126) or enalapril (n = 127). Conventional treatment for HF, including the use of other vasodilators, was continued in both groups. Follow-up averaged 188 days (range, 1 day to 20 months). The crude mortality at the end of 6 months (primary end point) was 26 % in the enalapril group and 44 % in the placebo group: a reduction of 40 % (P = 0.002). Mortality was reduced by 31 % at 1 year (P = 0.001). By the end of the study, there had been 68 deaths in the placebo group and 50 in the enalapril group: a reduction of 27 % (P = 0.003). The entire reduction in total mortality was found to be among patients with progressive HF (a reduction of 50 %), whereas no difference was seen in the incidence of sudden cardiac death. A significant improvement in NYHA classification was observed in the enalapril group, together with a reduction in heart size and a reduced requirement for other medication for HF.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


