Abnormalities in Calcium Cycling in Heart Failure
Judith Karen Gwathmey
Federica del Monte
Roger Joseph Hajjar
The major abnormality in the failing heart is loss of contractility. The seemingly logical corollary that an increase in intracellular calcium concentration would benefit patients with chronic heart failure stimulated the development of inotropic agents. The premise was that myocardial contractility would be restored by increasing myocardial calcium concentration.
Supporting this concept was the observation that adaptive/maladaptive changes that occurred in failing myocardium, such as altered sarcoplasmic reticulum (SR) calcium ATPase activity, isoform replacement of proteins involved in adenosine triphosphate (ATP) supply and demand, as well as isoform shifts in contractile proteins, impacted myofilament activation and calcium concentration/mobilization. The proteins essential for calcium sequestration and release, myofilament activation, and energy production are now known to not only be altered but also to have functional consequences. In this chapter we will address the question of whether changes in structure and/or function of calcium cycling proteins alone or in combination with deprivation of energy supply (e.g., ATP and phosphocreatine [PCr]), and/or changes at the level of the myofilaments, and/or accumulation of metabolites are sufficient to alter calcium-regulated contractile activation and contribute to impaired contractility in the failing human heart.
Excitation-Contraction Coupling in the Heart
Figure 5-1 demonstrates a simplistic schematic of excitation-contraction in the heart. The heart rhythmically goes through each beat, which is governed by the contraction and relaxation cycle of individual cardiac myocytes and the heart as a whole. The process begins with electrical signals on the membrane of the cardiac cell that eventually end in activation of the myofilaments and cross-bridge cycling, resulting in force production (1). The cardiac action potential is produced by the coordinated interaction of several ion channels that transduce physiological signals within and between cardiac myocytes.
Action potential conduction in the heart depends on ion conductance through specific voltage-gated ion channels that mediate rapid, voltage-dependent changes in ion permeability, which causes a change in the membrane potential. In the human heart, depolarization of the cell membrane leads to the opening of voltage-gated L-type calcium channels, located in the T-tubular regions of the cardiac myocyte, allowing the trans-sarcolemmal influx of a small amount of calcium into the cell (Fig. 5-1) (1,2,3,4,5,6,7,8). The L-type calcium channels are located in close proximity to the calcium release channels, known as ryanodine receptors, which are located on the SR (the major calcium-storing organelle in the cardiac myocyte). Calcium entering the cells through a single L-type calcium channel triggers the opening of one or a cluster of ryanodine receptors, resulting in the local release of calcium from the SR (1,2,3,4,5,8). During membrane depolarization, a large number of L-type calcium channels are opened, resulting in a large release of calcium from the SR through the ryanodine receptors, raising cytosolic calcium from 0.1–0.2 μmol/L to 2–10 μmol/L.
The calcium spark has been identified as a functional calcium signaling and releasing unit (4). Functionally, calcium sparks are calcium releases from the SR through the opening of the ryanodine receptors. Calcium sparks are depicted and measured by their morphology and the
frequency of occurrence. Morphology of the calcium sparks refers to the amplitude, width, and duration of the spark. The more active the calcium release channels are, the more calcium is released from the SR. Therefore, adaptive modifications that change ryanodine receptor activity or the modulators that regulate ryanodine receptor activity will have an impact on the size and frequency of calcium sparks and the subsequent release of calcium into the cytosol.
frequency of occurrence. Morphology of the calcium sparks refers to the amplitude, width, and duration of the spark. The more active the calcium release channels are, the more calcium is released from the SR. Therefore, adaptive modifications that change ryanodine receptor activity or the modulators that regulate ryanodine receptor activity will have an impact on the size and frequency of calcium sparks and the subsequent release of calcium into the cytosol.
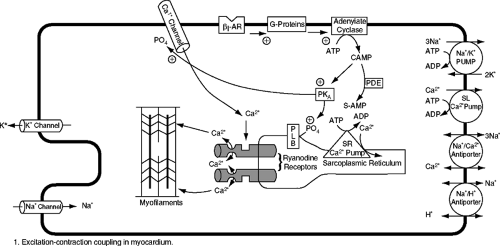 Figure 5-1 In the scheme of excitation-contraction coupling, there are two competing mechanisms for removing calcium from the cytoplasm: SERCA2a and the sodium-calcium exchanger. SERCA2a is inhibited by phospholamban when this protein is unphosphorylated. Phosphorylation of phospholamban through protein kinase A, calmodulin, and protein kinase C is balanced by dephosphorylation through protein phosphatases. (From Gwathmey JK, Hajjar RJ. The complexity of simplicity: the pathopysiology of heart failure. Hype or hope? Res Phys. 1993;39(4):45–49 , with permission.) |
Calcium content of the SR also regulates calcium sparks. SR luminal calcium concentration plays a critical role in regulating ryanodine receptors by increasing the activity of the receptors and by sensitizing the threshold of receptor activation by signaling between L-type calcium channels and ryanodine receptors. Calcium sparks can be directly triggered by the influx of calcium through the L-type calcium channels. Thus, the kinetics, fidelity, and stoichiometry of coupling between L-type calcium channels and ryanodine receptors may play a critical role in the determination of signal transduction during excitation-contraction coupling. Although the number of calcium channels in failing human hearts has been reported to not be changed, the ryanodine receptors have been reported to be leaky, suggesting possible uncoupling or structural changes (9,10,11,12,13,14,15). The distance of the cleft between the cell membrane and the SR membrane, which is roughly 12 nm in normal cardiac myocytes, is thought to be critical for proper coupling and signal transduction between the L-type calcium channels and the ryanodine receptors. Expansion of the distance between L-type calcium channels and ryanodine receptors can therefore decrease or abolish the signaling reliability. This potential structural uncoupling may therefore impact the amount of calcium released from the SR. Furthermore, the amount of calcium stored in the SR may be reduced, thereby resulting in less calcium being available upon release for contractile activation at higher rates of contraction.
The rise of calcium in the myofibrillar space in turn results in cross-bridge formation and contraction (16,17). The contractile elements consists of an array of thick and thin filaments. The thick filaments consist mainly of myosin heavy chains (MHCs) and two pairs of light chains (16). Each myosin head has an ATP-binding area in close proximity to myosin ATPase, which cleaves ATP. Two MHC isoforms exist in human myocardium, alpha- and beta-MHC. In human ventricular myocardium only 10% to 20% is alpha-MHC, while the beta-MHC is more abundant (18). The thin filaments are composed of actin and the troponin complex. These regulatory proteins are carried on a long, helical molecule called tropomyosin. The troponin complex is made up of troponin C, troponin 1, and troponin T. Troponin C is the calcium-binding protein. The strength of the bond linking troponin 1 (an inhibitory protein) and actin varies depending on the intracellular calcium concentration.
When cytosolic calcium is in the nanomolar range, the tropomyosin-troponin complex is positioned in such a way that the myosin heads cannot interact with actin. When cytosolic calcium increases to micromolar ranges, actin binds to troponin C, strengthening the bond between troponin C and troponin 1 and weakening the bond linking troponin 1 to actin. This leads to a conformational change of the tropomyosin-troponin C complex that allows the myosin head to interact with actin (Fig. 5-2).
When calcium binds to troponin C, it exposes the active site on the thin filament, permitting the myosin head to bind weakly to actin filaments. Subsequent ATP hydrolysis allows a strong binding of the myosin head to actin, which is followed by a power stroke that produces the force of contraction (16). Relaxation occurs when ATP binds to the myosin head, causing a dissociation of the thin from the thick filaments. Calcium is detached from troponin C and is resequestered into the SR by the calcium ATPase pump (SERCA2a) and is extruded from the cell by the sarcolemmal sodium-calcium exchanger.
When calcium binds to troponin C, it exposes the active site on the thin filament, permitting the myosin head to bind weakly to actin filaments. Subsequent ATP hydrolysis allows a strong binding of the myosin head to actin, which is followed by a power stroke that produces the force of contraction (16). Relaxation occurs when ATP binds to the myosin head, causing a dissociation of the thin from the thick filaments. Calcium is detached from troponin C and is resequestered into the SR by the calcium ATPase pump (SERCA2a) and is extruded from the cell by the sarcolemmal sodium-calcium exchanger.
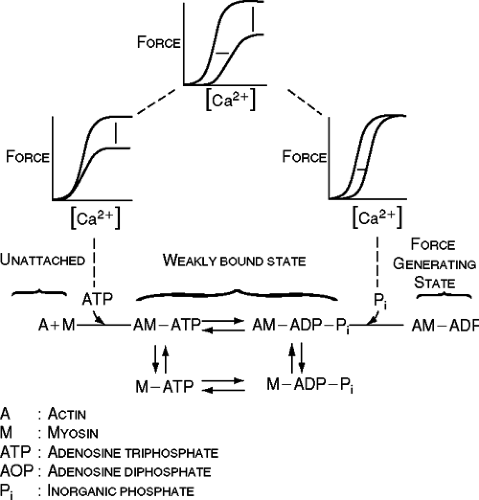 Figure 5-2 When cytosolic calcium increases to micromolar ranges, actin binds to troponin C, strengthening the bond between troponin C and troponin I and weakening the bond linking troponin I to actin. This leads to a conformational change of the tropomyosin-troponin C complex that allows the myosin head to interact with actin. When calcium binds to troponin C, it exposes the active site on the thin filament, permitting the myosin head to bind weakly to actin filaments. Subsequent ATP hydrolysis allows a strong binding of the myosin head to actin, which is followed by a power stroke that produces the force of contraction. Relaxation occurs when ATP binds to the myosin head, causing a dissociation of the thin from the thick filaments. Calcium is detached from troponin C and is resequestered into the SR by the calcium ATPase pump (SERCA2a) and is extruded from the cell by the sarcolemmal sodium-calcium exchanger. As shown in this figure, there are three types of changes that can decrease force production. (From Gwathmey JK, Hajjar RJ, Solaro RJ. Contractile deactivation and uncoupling of cross-bridges: effects of 2, 3-butanediene monoxine on mammalian myocardium. Circ Res. 1991;69: 1280–1292 , with permission.) |
As shown in Figure 5-2, there are three types of changes that can decrease force production (16). There can be a depression of maximal force (Fmax) that constitutes a change in the intracellular calcium concentration-force relationship. A decrease in Fmax can be the result of uncoupling of cross-bridges. However, it is known that the maximal calcium activated force in human failing myocardium is not diminished (2, 19,20,21). This has also been demonstrated in an avian model of heart failure (22,23). Accumulation of inorganic phosphate (as can be possibly seen as a result of ATP splitting) is known to affect the responsiveness of the contractile proteins in this manner (e.g., inorganic phosphate increases during hypoxia).
A decrease in intracellular calcium concentration, resulting in a decrease in force production along with an unaltered intracellular calcium concentration-force relationship, is another mechanism to be considered. As depicted in Figure 5-2, a rightward shift of the intracellular calcium concentration-force relationship can result from a decrease in the sensitivity of troponin C to calcium. A decrease in intracellular pH is known to produce this effect (e.g., during ischemia). Also, an increase in inorganic phosphate can result in a decrease in the sensitivity of the myofilaments to calcium, along with a decrease in maximal force. We will take a more in-depth look at the relationship between calcium availability, the myofilaments, and contractility.
The extent of contribution of the SR and the sodium-calcium exchanger to lowering cytosolic calcium concentrations varies among species. In humans, ∼75% of the calcium is removed from the cytosol by SERCA2a and ∼25% by the sodium-calcium exchanger. SERCA2a transports calcium back to the luminal space of the SR against a calcium gradient by an energy-dependent mechanism (one molecule of ATP is hydrolyzed for the transport of two molecules of calcium), where it binds to a calcium-buffering protein, calsequestrin.
The calcium-pumping activity of SERCA2a is regulated by phospholamban. In its unphosphorylated state, phospholamban inhibits the SR calcium-ATPase, whereas phosphorylation of phospholamban by cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) and by calcium-calmodulin-dependent PKA reverses this inhibition (24,25).
In the heart there are also a number of endogenous and exogenous factors that regulate the strength of contraction. Circulating and locally released catecholamines bind to myocardial β-1 and β-2 adrenoreceptors (26). β-3 receptors have been recently identified in the heart but their primary role appears to be an inhibitory effect on contractility. On the other hand, both β-1 and β-2 adrenoreceptors activate adenylate cyclase through stimulatory G proteins (Gs alpha), which results in the production of cAMP (Fig. 5-1). Binding of cAMP to the regulatory subunit of PKA triggers a conformational change that allows the catalytic subunits of the enzyme to dissociate and phosphorylate protein substrates. PKA phosphorylates (a) L-type calcium channels, resulting in increased calcium entry; (b) phospholamban, resulting in enhanced calcium uptake into the SR; and (c) troponin I, resulting in enhanced detachment of calcium from troponin C and decreased myofilament calcium sensitivity. The effects of PKA phosphorylation therefore increase the strength of the contraction while also enhancing relaxation.
Cardiac ryanodine receptors are also regulated by the beta-adrenergic receptor (beta-AR) signaling pathway. The ryanodine receptor has a delicate balance between PKA phosphorylation and phosphatases (i.e., protein phosphatase 1 [PP1] and 2 [PP2A]) (27). Activation of the sympathetic nervous system results in binding of norepinephrine to the beta-AR and an elevation of cAMP levels with resultant activation of PKA. In addition, there are other molecules such as FKB 12-6, which bind the L-type calcium channel
receptor to the ryanodine receptor and stabilize the interaction. Upon activation of the beta-AR, PP1 and PP2A levels in the ryanodine receptor decrease and the ryanodine receptors become phosphorylated. This pathologically increases calcium-dependent activation of the ryanodine calcium release channels and results in deletion of SR calcium stores. This results in an uncoupling of ryanodine receptors from each other, thereby reducing excitation-contraction coupling gain. This is important in that the ryanodine calcium release channels have been shown to become leaky in failing human hearts (9,12,13,14).
receptor to the ryanodine receptor and stabilize the interaction. Upon activation of the beta-AR, PP1 and PP2A levels in the ryanodine receptor decrease and the ryanodine receptors become phosphorylated. This pathologically increases calcium-dependent activation of the ryanodine calcium release channels and results in deletion of SR calcium stores. This results in an uncoupling of ryanodine receptors from each other, thereby reducing excitation-contraction coupling gain. This is important in that the ryanodine calcium release channels have been shown to become leaky in failing human hearts (9,12,13,14).
One can readily understand how depleted SR calcium stores can result in reduced contractile activation, a negative force-interval relationship (i.e., negative treppe), thereby negatively impacting myocardial contractility (Fig. 5-3) (12,14). The force-frequency relationship significantly contributes to the beat-to-beat regulation of the contractile strength in the heart. In a normal heart, increasing heart rate results in enhanced trans-sarcolemmal calcium influx, resulting in more calcium being stored in the SR for subsequent release and stronger contraction. However, in the failing human heart a negative force-interval relationship has been reported (28,29,30,31,32,33,34). At higher rates of contraction there is a decline in force as well as an increase in diastolic force (Fig. 5-3) (28,29,30,31,32,33,34). This negative treppe is associated with a dramatic increase in diastolic calcium concentrations and a reduction in the amplitude of the peak calcium transient. Also, there is evidence that at higher contraction rates there is likely an accumulation of metabolites (e.g., inorganic phosphate) or possibly a change in intracellular pH; this appears to result in a rightward shift on the calcium axis, resulting in diminished force for any given intracellular calcium concentration in the cell (Fig. 5-4) (29).
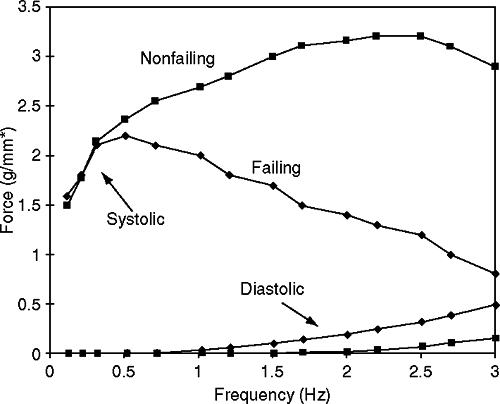 Figure 5-3 Force-interval relationship. There is no difference in the force of contraction in isolated muscle preparations from nonfailing and failing human hearts at slower stimulation rates. However, at higher rates of stimulation there is a decrease in the force of contraction seen in muscles isolated from failing human hearts unlike the continued increase in force noted in muscles from nonfailing human hearts. Also noted in muscles from failing human hearts associated with the decline in systolic force of contraction was an increase in diastolic force at higher rates of contraction. This observation has been shown to be associated with a rise in intracellular diastolic calcium concentration. (From Schmidt U, Hajjar RJ, Gwathmey JK. The force-interval relationship in human myocardium. J Card Fail. 1995;1(4):311–321 , with permission.) |
As demonstrated in Figure 5-4, with delays after high rates of stimulation before tetanization in isolated muscle strips from the hearts of patients with heart failure, there is an increase in calcium-activated force with no change in extracellular calcium concentration. Similarly, it was demonstrated that despite having the same intracellular calcium concentration as a muscle subjected to lower rates of contraction, muscles stimulated at higher rates of contraction and then tetanized generated less force. This dramatic example suggests that changes in myofilament calcium activation and contractility may change in a dynamic manner, possibly on a beat-to-beat basis, and might partly explain why, at lower rates of stimulation, peak twitch force is similar for muscle strips from nonfailing and failing human hearts (Fig. 5-3).
Hypotheses for Decreased Contractile Performance in the Failing Heart
Calcium Mobilization Versus Availability: Impact on Contractile Performance
It has been proposed that the diminished contractility seen in the failing heart is due to diminished intracellular calcium. Using the calcium indicator Indo in isolated myocytes from failing human hearts, it was reported that peak SR calcium release was 1 μmol/L, a value similar to that observed for normal mammalian myocardium (35). Furthermore, a significantly higher resting calcium concentration has been demonstrated, particularly with increased rates of stimulation and contraction. These experimental findings have been confirmed in multicellular preparations using aequorin and Fura-2 (28,29).
Experiments using aequorin have reported an increased diastolic calcium concentration associated with an increase in diastolic force in multicellular preparations similar to that reported in isolated cardiac myocytes (28,29). Only at higher stimulation rates was there a decrease in peak cytosolic calcium concentration detected with aequorin. Del Monte et al. also reported that there was a decrease in peak systolic calcium at high stimulation rates, with an associated elevated diastolic calcium concentration in cardiac myocytes from failing human hearts (28). The peak calcium concentration at basal stimulation rates for myocytes from nonfailing human hearts versus myocytes from failing human hearts was 551 ± 32 nmol/L, versus 508 ± 25 (p >0.05) nmol/L, respectively. As reported with aequorin in multicellular preparations, with higher frequencies of contraction there was a disproportionate rise in the diastolic calcium concentration, a reduced peak systolic calcium transient, and slowed relaxation (29,36) (Fig. 5-5). Despite the controversy as to whether or not the peak of the calcium transient is diminished at lower rates of contraction, one must take into consideration the effect of a longer duration of calcium availability, which has been demonstrated with all calcium indicators (Figs. 5-5 and 5-6)
(28,36). Time course and duration of calcium availability can be key factors in determining “apparent myofilament calcium sensitivity and calcium responsiveness” (37).
(28,36). Time course and duration of calcium availability can be key factors in determining “apparent myofilament calcium sensitivity and calcium responsiveness” (37).
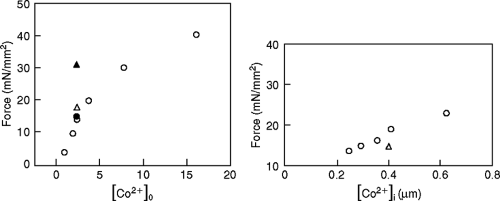 Figure 5-4 Left panel steady-state force-calcium relationship in myopathic tissue in the presence of increasing extracellular calcium concentrations at a basal stimulation rate of 0.33 Hz (o). Tetanization basal frequency of 1 Hz (•) without delay; 5-second delay (Δ); 20-second delay (▲). Right panel is the steady-state force-intracellular calcium relationship at varying intracellular calcium concentrations at 0.33 Hz (o) compared to basal frequency of 1 Hz in 2.5-mmol/L extracellular calcium without delay (Δ). The left panel demonstrates that with delays, despite no change in intracellular calcium, the force increases. The right panel shows that despite having the same intracellular calcium at 1 Hz as at 0.33 Hz, the force is less. These data suggest a change in myofilament calcium responsiveness. (From Gwathmey JK, Slawsky MT, Hajjar RJ, et al. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J Clin Invest. 1990;85(5):1599–1628 , with permission.) |
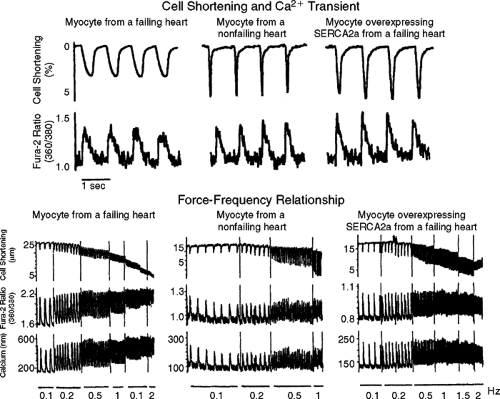 Figure 5-5 The top panel demonstrates cell shortening and associated calcium transient in an isolated cardiac myocyte from a nonfailing human heart and a failing human heart. The far right panel demonstrates how transfection with SERCA2a restores cell shortening and abbreviates the time course of the calcium transient. The lower panels demonstrate the impact of increasing the frequency of shortening in an isolated myocyte from nonfailing and failing hearts. Myocyte overexpressing SERCA2a has a more normal force interval relationship. There was a significant reduction in the increase in diastolic calcium concentrations and reduced shortening noted in a myocyte isolated from a failing human heart with SERCA2a transfection. The same hearts were shown to have a significant reduction in SERCA2a protein content. (From del Monte F, Harding S, Schmidt U, et al. Restoration of contractile function in isolated cardiomyocytes from failing human hearts by gene transfer of SERCA2a. Circulation 1999; 100(23):2308–2313 , with permission.) |
Myofilament Calcium Responsiveness and Heart Failure
Contractility is determined not only by the amount of calcium that is available in the cytosol, but also by the duration and time course of calcium availability and the duration of troponin C-calcium interaction (5,37). The first report of intracellular calcium signals in nonfailing and failing human myocardium demonstrated a slowed time course to the decay from the peak calcium transient to diastolic levels (Fig. 5-6) (36). An elevated diastolic calcium concentration was also clearly demonstrated (28,29,36). These first images that captured calcium handling in nonfailing and failing human myocardium challenged the then-current dogma that more calcium was needed in the failing heart. They also raised the question of whether better mobilization of calcium was needed, as opposed to the use of inotropes that acted by increasing intracellular calcium concentrations.
In response to this unexpected observation, many investigators attempted to make inferences about changes in calcium responsiveness and force of contraction by using the peak twitch force-peak light or calcium relationship. This was then extended to the use of fluorescence indicators in isolated myocytes and became the peak fluorescence-peak shortening relationship. We, as well as other investigators, have discussed the limitations of the use of this technique and the fact that the relationship can be artifactually shifted by alterations in the duration of the calcium transient, time course of contraction, as well as changes in the cross-bridge cycling kinetics (17). To make this point clearer and to demonstrate the impact of changes in the time course of calcium mobilization and time course of contraction on contractility, we performed several studies using intracellular calcium signals as well as skinned fiber preparations. The advantage of using skinned fibers is that the exact amount of calcium delivered to the contractile elements is known and can be studied under steady-state conditions.
We compared peak twitch force-peak calcium relationships to skinned fiber force-calcium relationships in muscles from the same experimental animal. We established two models of disease: pressure-overload hypertrophy (POH) and hyperthyroidism in the ferret. We were motivated to do this for several reasons. With ventricular POH, isolated muscles demonstrate an isometric twitch and associated intracellular calcium transient that are prolonged (38,39,40). In contrast, ferrets made hyperthyroid by injection of thyroxine have abbreviated time courses of the isometric twitch and intracellular calcium (41). In both situations (POH and hyperthyroidism) there are no significant differences in diastolic or peak intracellular calcium concentration. These two models, therefore, provide an opportunity to study the effects of changes in time course of the intracellular calcium transient and isometric twitch in the presence of similar amounts of peak and diastolic intracellular calcium concentrations. Therefore, we had a relatively simple system to investigate the effect of time course changes of the calcium transient and contraction on myocardial contractility and the apparent force-calcium relationship.
As mentioned earlier, there are two key players in the calcium signal reported with calcium indicators: (a) troponin C Ca2+ binding affinity and (b) SR function. Troponin C and the SR are the most important calcium sinks in myocardial cells. Troponin C binds Ca2+ immediately after the ion enters the myofibrillar space. Within 20 milliseconds in frog skeletal muscle at 0°C, troponin C is at least 90% saturated (42). Force does not begin to rise until after 10 milliseconds. Therefore, if the affinity of troponin C is altered it will affect the time course as well as amplitude of the calcium signal. Similar to placing a low-pass filter in an electronic circuit, as the affinity increases, the time course will become longer and the peak shorter. If the affinity decreases, the calcium signal will be briefer and taller. Therefore, the amplitude of the calcium transient is determined by the rate at which the SR releases
calcium and the rate at which troponin C binds the free calcium.
calcium and the rate at which troponin C binds the free calcium.
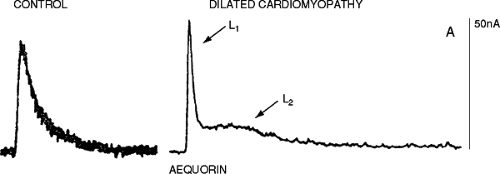 Figure 5-6 Intracellular calcium transients from isolated muscles from nonfailing and failing human hearts loaded with aequorin, a bioluminescent protein. The left panel is calcium transient from a nonfailing human heart and the right panel is from a patient who had undergone cardiac transplantation for dilated cardiomypathy. (From Gwathmey JK, Copelas L, Mackinnon R, et al. Abnormal intracellular cacium handling in myocardium from patients with end-stage heart failure. Circ Res. 1987;61:70–76 , with permission.) |
The time course of the intracellular calcium signal is therefore regulated by the balance between the release of calcium and its uptake by the SR. Thus, one would anticipate that interventions that increase the affinity of troponin C to calcium would decrease the amplitude of the intracellular calcium signal. They would also be expected to decrease the decline of intracellular calcium signal since calcium would be released more slowly from troponin C. On the other hand, a decrease in the affinity of troponin C to calcium would result in an elevated peak intracellular calcium concentration and a shorter time course. Agents that increase the rate of calcium uptake by the SR would be expected to shorten the time course of the calcium signal without affecting the peak intracellular calcium concentration, and agents that decrease the rate of calcium uptake by the SR would be expected to lengthen the time course of intracellular calcium transient without affecting the peak intracellular calcium concentration.
It is important to note that excluded in our discussions are the effects on the time course or amplitude of the calcium signal of cellular extrusion mechanisms (e.g., the sodium-calcium exchanger, as well as additional calcium buffers such as mitochondria). It is thought that the effects are relatively small compared to the key players and that these factors are less subject to large kinetic changes.
As shown in Figures 5-7 and 5-8, we can demonstrate shifts in the apparent calcium sensitivity for force calcium activation in the absence of a true change in myofilament calcium sensitivity. In the case of POH, one obtains a higher force for any given level of extracellular calcium. In contrast with hyperthyroidism, decreasing the time course of the intracellular calcium transient resulted in a rightward shift on the calcium axis (i.e., an apparent decrease in the sensitivity of the myofilaments despite having with no change in sensitivity of the myofilaments). Therefore, investigators must use caution when drawing conclusions regarding myocardial contractility and its relationship to intracellular calcium concentrations.
Our examples demonstrate that a slower calcium transient as well as a faster calcium transient alone can alter the apparent force-calcium relationship and myocardial contractility in a predictable direction (Fig. 5-9) (37,38,39,40,41). It is important to note that in skinned fibers from the same hearts with pressure overload or hyperthyroidism there is no change in the force-calcium relationship (37). This is similar to our observations in myocardial fibers from failing and nonfailing human hearts (Fig. 5-10) (20,21). In skinned fibers from nonfailing and failing human hearts, no differences in myofilament calcium sensitivity or maximal calcium activated force were detected (20,21). This observation has been confirmed by us at the level of a single isolated myocyte (Fig. 5-11) (unpublished data). These observations are identical to reports in an avian model of heart failure that we have demonstrated mimics subcellular as well as genomic changes seen in the failing human heart (22,23). However, if multicellular muscle preparations were challenged with changes in intracellular pH, phosphate, PKA, PKC, and cAMP, differences were
detected in the force-calcium relationship between fibers from nonfailing and failing human hearts, similar to our observations in an avian model of heart failure (20,21,22,23,43).
detected in the force-calcium relationship between fibers from nonfailing and failing human hearts, similar to our observations in an avian model of heart failure (20,21,22,23,43).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


