Familial dilated cardiomyopathy is a major cause of advanced heart failure and heart transplantation. In most families, the disease-causing mutation is unknown, and relatives should therefore undergo periodic screening to facilitate early diagnosis and therapy. In the present study, we describe a novel titin truncation mutation causing adult-onset familial dilated cardiomyopathy in an Israeli Arab family. The family members underwent physical examination, electrocardiography, and Doppler echocardiography. Linkage to candidate loci was performed, followed by gene sequencing. We identified 13 clinically affected family members (8 men and 5 women, mean age 47 ± 12 years). Compared with their healthy first-degree relatives, the affected relatives had a larger end-diastolic left ventricular dimension (60 ± 10 vs 49 ± 4 mm, p <0.001), lower ejection fraction (43 ± 11% vs 60 ± 6%, p <0.001), and markedly higher end-systolic volume indexes but no difference in wall thickness or diastolic function. The linkage studies or direct sequencing excluded LMNA, MYH7, TNNT2, TNNI3, SCN5A, DES, SGCD, ACTC, PLN, and MYH6 but established linkage to the TTN locus at chromosome 2q31, yielding a maximum (2-point) LOD score of 3.44. Sequence analysis identified an insertion (c.58880insA), causing protein truncation after 19,628 amino acids (p.S19628IfsX1). No founder effect was found among the Israeli Arabs. In conclusion, titin is a giant protein with a key role in sarcomere assembly, force transmission, and maintenance of resting tension. Although some mutations result in skeletal myopathy, others cause isolated, maturity-onset cardiomyopathy.
Idiopathic dilated cardiomyopathy (IDC) is a major cause of heart failure and, hence, heart transplantation in young adults. Clinical and genetic studies of families with members affected by IDC suggest a familial etiology in approximately 30% of cases. The mode of inheritance is generally autosomal dominant, although X-linked and autosomal recessive transmission have also been described. Mutations causing IDC are found in a wide spectrum of genes encoding for proteins involved in force generation, force transmission, metabolism, calcium homeostasis, and transcription regulation. A cardinal element of the sarcomere is the giant protein titin, which physically connects myosin fibers to actin polymers attached to the Z-line. Titin has multiple biologic functions, including maintenance of resting tension and elasticity (by way of a series of spring elements), stabilization of contractile proteins, and force transmission. Mutations in titin have been described in several families inherited as an autosomal dominant trait. In the present study, we identified a novel titin mutation in a Muslim Arab family, with adult-onset familial dilated cardiomyopathy (FDC).
Methods
The study conformed to the principles outlined in the Declaration of Helsinki and was approved by the institutional review board, Sheba Medical Center, and the Genetic Committee of the Ministry of Health, Israel. We identified a large family, designated D4CH, with maturity-onset FDC. All participants or their legal representatives provided informed consent before inclusion in the present study. The patients and family members were examined by a dedicated team from the heart failure service. After reviewing the medical records, we clinically evaluated and extracted DNA from 43 adult family members to perform linkage analysis.
All Doppler echocardiographic studies were performed using Vivid 7 or mobile Vivid I devices (General Electric, Tirat Carmel, Israel) using a 2/5-MHz transducer and were recorded digitally for additional off-line analysis. Blood was drawn into ethylenediaminetetraacetic acid (EDTA)-containing tubes and stored at 4°C for ≤24 hours.
DNA was extracted using a Puregene DNA isolation kit (Gentra Systems, Minneapolis, Minnesota) and diluted to 200 ng/μl. Initially, we studied linkage to previously established FDC-causing genes using polymorphic short tandem repeat markers adjacent to the locus of interest. Nucleic acid sequences were obtained from GenBank or Sanger ( www.ensembl.org ). Primers were generated with primer-3 software and are available on request. Allele lengths were determined by electrophoresis with an ABI-3100 Genetic Analyzer and Genotyper software (ABI PRISM ® 3100 Genetic Analyzer, Applied Biosystems, California). All exons of the TTN gene known to be expressed in cardiac muscle were amplified using polymerase chain reaction and directly sequenced using a Big Dye Terminator DNA sequencing kit on a 3100 Avant Genetic Analyzer (PE Applied Biosystems, California), according to a previously described protocol. The TTN c.58880insA mutation in exon 326 ( AJ277892 , NM003319 ) was first confirmed by subcloning the polymerase chain reaction product into a pTRE2.1-TOPO vector (Invitrogen, Invitrogen Ltd, UK) to separate the alleles containing 6 or 7 adenine residues. Family members were genotyped through the elution waveform of a 92-bp fragment from exon 326, using an ABI-3100 Genetic Analyzer.
Affection status was determined on the basis of the consensus document ; however, in echocardiographically borderline cases, we also considered a pre-existing clinical diagnosis of cardiomyopathy. The LOD score was calculated using the LINKAGE, version 5.1 package (BIMAS, Salt Lake City, Utah). Statistical comparisons were performed using Student’s t test or Fisher’s exact tests, with p <0.05 as statistically significant.
Results
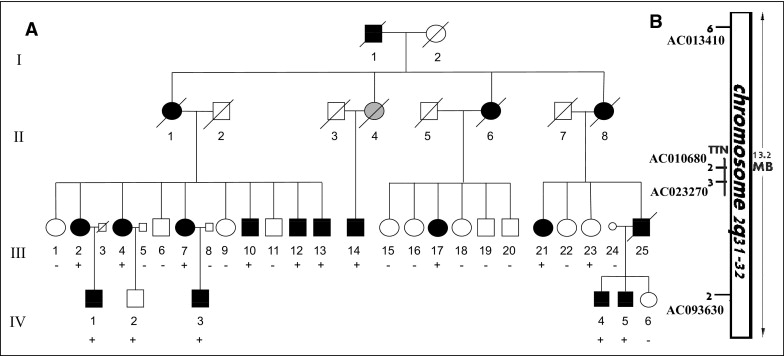
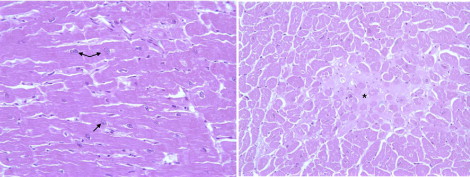
The family with late-onset FDC is a Muslim Arab family residing in Galilee, Israel ( Figure 1 and Table 1 ). The proband, a 37-year-old man (III-13; Figure 1 and Table 1 ), was referred for evaluation for heart transplantation because of refractory heart failure. His brother (III-10; Figure 1 and Table 1 ) had undergone heart transplantation elsewhere at the age of 41 years. Their mother (II-1; Figure 1 ) had died at 67 years old from severe congestive heart failure. A cousin (III-25; Figure 1 ) had died at 49 years old while awaiting a heart transplant. The family included several members previously diagnosed with symptomatic heart failure and/or IDC. We performed a clinical evaluation and extracted DNA to perform linkage analysis in 43 family members. Of these, 13 were affected by cardiomyopathy ( Table 1 ), and 30 first-degree family members had no clinical evidence of the disease. Except for the heart transplant recipients, 6 of the 43 family members had symptomatic heart failure. There was no clinical evidence or family history of skeletal myopathy, conduction system disease, ventricular arrhythmia, or sudden cardiac death. The pathologic findings from an explanted heart demonstrated nonspecific cardiomyopathic changes, including mild myocyte hypertrophy and interstitial fibrosis ( Figure 2 ) .
| Subject Code | Age (yr) | Gender | Heart Failure | Heart Transplantation | LV End-Diastolic Diameter (mm) | LV End-Systolic Diameter (mm) | Ventricular Septum (mm) | LVEF (%) | Left Atrium Diameter (mm) | ECG Findings |
|---|---|---|---|---|---|---|---|---|---|---|
| III-2 | 59 | Female | Yes | No | 57 | 38 | 12 | 52 | 42 | Normal |
| III-4 | 65 | Female | Yes | No | 70 | 59 | 9 | 48 | 56 | First atrioventricular block, left bundle brunch block, P mitrale, poor R wave progression |
| III-7 | 54 | Female | No | No | 48 | 38 | 12 | 42 | 38 | Poor R wave progression |
| III-10 | 41 | Male | Yes | Yes | — | — | — | — | — | — |
| III-12 | 51 | Male | Yes | No | 58 | 47 | 10 | 43 | 44 | Poor R wave progression |
| III-13 | 37 | Male | Yes | Yes | 74 | 67 | 10 | 20 | 61 | First atrioventricular block, intraventricular conduction defect, P mitrale, poor R wave progression |
| III-14 | 58 | Male | Yes | Yes | 76 | 71 | 9 | 20 | 44 | Pacemaker, P mitrale |
| III-17 | 36 | Female | Yes | No | 56 | 42 | 9 | 50 | 39 | Normal |
| III-21 | 54 | Female | Yes | No | 64 | 54 | 8 | 45 | 41 | Left bundle brunch block, left axis deviation, poor R wave progression |
| IV-1 | 39 | Male | Yes | No | 51 | 37 | 11 | 41 | 40 | Poor R wave progression |
| IV-3 | 27 | Male | No | No | 60 | 48 | 9 | 47 | 37 | Poor R wave progression |
| IV-4 | 48 | Male | No | No | 53 | 38 | 9 | 45 | 37 | Right bundle brunch block, left axis deviation |
| IV-5 | 38 | Male | No | No | 47 | 36 | 11 | 47 | 38 | Left axis deviation, poor R wave progression |
The pedigree and clinical data ( Figure 1 and Table 1 ) were compatible, with autosomal dominant adult-onset FDC with age-dependent penetrance. Thus, healthy subjects were defined as “nonaffected” only after 40 years of age. Studies of linkage to candidate loci excluded 8 known FDC-causing genes ( MYH7, LMNA, TNNT2, TNNI3, SCN5A, DES, SGCD , and MYH6 ) because of a complete lack of segregation with the phenotype. Direct sequencing did not identify a mutation in the ACTC and PLN genes. An examination of the intragenic polymorphic marker located at AC010680 , in intron 244 of TTN gene (chromosome 2q31) revealed segregation in all the affected family members. Additional testing with additional polymorphic markers surrounding the TTN gene defined a common haplotype in all the affected subjects, yielding a maximum 2-point LOD score of 3.44 at θ = 0 with a marker located at AC013410 ( Figure 1 ), assuming 90% penetrance and 0.0003 disease allele prevalence. Sequence analysis of the TTN gene identified a novel mutation in exon 326. In this mutation, an adenine insertion at position 58,880 (c.58880insA) causes a frameshift, creating a stop codon and protein truncation after 19,628 amino acids (p.S19628IfsX1, Figure 3 ) . The mutation was confirmed by subcloning the alleles into an expression vector ( Figure 3 ) and by a fragment elution waveform ( Figure 3 ) and was not found in 200 healthy controls, including 100 local subjects of Arab origin.
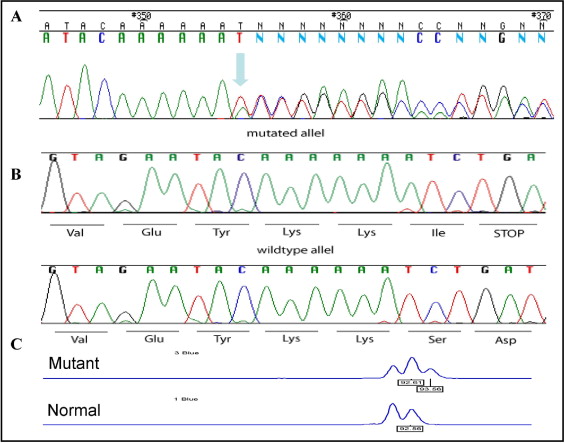
Genotyping of the family members identified the TTN c.58880insA mutation in all 13 patients with FDC. Of the 30 healthy relatives, the mutation was present in 6 (3 men and 3 women, aged 27 to 48 years) and in 24 (11 men and 13 women, aged 29 to 56 years) the genotype was negative. We then examined whether this mutation could constitute a founder mutation among Israeli Arabs that would explain a portion of local IDC. The mutation was not found in 2 other FDC families of the same ethnic origin, including a family from a neighboring village.
To better delineate the disease phenotype, we compared the electrocardiographic and echocardiographic measures of the clinically affected subjects with their healthy mutation-negative first-degree relatives. Electrocardiography of the affected subjects often exhibited left axis deviation, complete left bundle branch block, or intraventricular conduction defect. The average PR interval was 188 ± 4 ms in the affected subjects compared to 160 ± 20 ms in controls (p <0.01). In addition, poor R progression was common in the patients (8 of 11 patients with a nonpaced electrocardiographic study compared to 6 of 24 controls, p <0.01). No significant differences were seen in electrocardiographic voltage, P or T wave morphology. On the Doppler echocardiogram, affected subjects had larger ventricles and markedly impaired systolic function, including left ventricular shortening fraction, ejection fraction, and end-systolic dimension ( Table 2 ). The right ventricular contraction was also affected. No significant differences were seen in the wall thickness or indexes of diastolic function. All healthy carriers had normal echocardiographic and electrocardiographic findings.
| Parameter | Affected (n = 12) ⁎ | Unaffected (n = 24) | p Value |
|---|---|---|---|
| Gender | 0.48 | ||
| Male | 7 | 11 | |
| Female | 5 | 13 | |
| Age (years) | 47 ± 11 | 43 ± 7 | 0.22 |
| Weight (kg) | 84 ± 16 | 87 ± 13 | 0.63 |
| Height (cm) | 170 ± 10 | 169 ± 10 | 0.74 |
| Body surface area (kg/m 2 ) | 2.0 ± 0.2 | 2.0 ± 0.2 | 0.73 |
| Heart rate (beats/min) | 78 ± 10 | 78 ± 12 | 0.99 |
| Left ventricular end-diastolic diameter (mm) | 60 ± 10 | 49 ± 4 | <0.001 |
| Left ventricular end-systolic diameter (mm) | 48 ± 12 | 30 ± 5 | <0.001 |
| Ventricular septum (mm) | 10 ± 1 | 10 ± 1 | 0.79 |
| Posterior wall (mm) | 10 ± 1 | 10 ± 1 | 0.92 |
| Shortening fraction (%) | 20 ± 8 | 39 ± 7 | <0.001 |
| Left ventricular mass | 289 ± 85 | 203 ± 52 | <0.001 |
| Left ventricular mass index | 148 ± 52 | 100 ± 20 | <0.001 |
| Left ventricular end-diastolic volume (cm 3 ) | 147 ± 38 | 104 ± 30 | 0.001 |
| Left ventricular end-systolic volume (cm 3 ) | 76 ± 21 | 42 ± 16 | <0.001 |
| Left ventricular ejection fraction (%) | 43 ± 11 | 60 ± 6 | <0.001 |
| Left ventricular end-diastolic volume index | 73 ± 19 | 51 ± 12 | <0.001 |
| Left ventricular end-systolic volume index | 38 ± 10 | 21 ± 7 | <0.001 |
| Left atrium diameter (mm) | 43 ± 8 | 39 ± 3 | 0.02 |
| Left atrium volume (4C) | 65 ± 41 | 48 ± 11 | 0.06 |
| Left atrium volume index | 33 ± 22 | 24 ± 5 | 0.06 |
| Left atrium area (cm 2 ) | 22 ± 8 | 17 ± 2 | <0.01 |
| Mitral valve E (cm/s) | 68 ± 20 | 80 ± 19 | 0.09 |
| Mitral valve A (cm/s) | 59 ± 23 | 66 ± 26 | 0.39 |
| Mitral valve E/A | 1.6 ± 0.3 | 1.3 ± 0.4 | 0.35 |
| Mitral valve E deceleration time (ms) | 170 ± 32 | 187 ± 53 | 0.31 |
| E/E′ | 8 ± 3 | 8 ± 2 | 0.61 |
| Right ventricular thickness (mm) | 5.2 ± 1.0 | 4.9 ± 0.7 | 0.31 |
| Right ventricular diastolic area (cm 2 ) | 16 ± 4 | 16 ± 2 | 0.53 |
| Right ventricular systolic area (cm 2 ) | 10 ± 2 | 9 ± 2 | 0.17 |
| Right ventricular fractional area change (%) | 40 ± 6 | 45 ± 5 | 0.02 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


