Fig. 10.1
Twelve-lead ECG showing peaked T waves. The QT interval is prolonged. Paper speed 25 mm/s, 10 mm/mV
A 24-h tape and an exercise test were performed and confirmed a persistently prolonged QT interval. The QTc remained fixed with no adaptation to change in rate (Fig. 10.2).
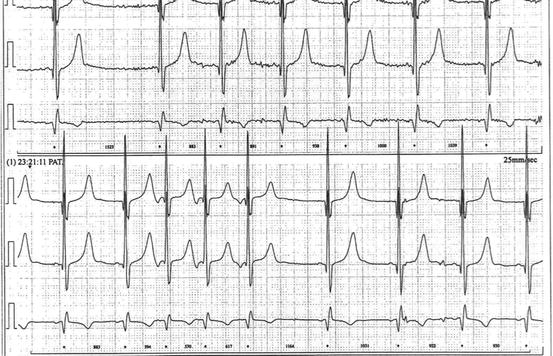
Fig. 10.2
Section of 24-h recording showing no adaptation to rate change in QT interval
The patient had been informed about the suspected diagnosis and had been given lifestyle advice that included limiting exercise. He was advised to stop playing 5-a-side football. He was commenced on beta-blocker therapy with bisoprolol.
Blood analysis had been said to have been done early on in his screening, but blood was taken in the inherited cardiac conditions clinic for genetic analysis and also sent for urea and electrolytes and thyroid function.
Surprisingly, his blood test was returned reporting very abnormal parameters with hypomagnesaemia, hypocalcaemia and hyperphosphatemia and high urea and creatinine (Table 10.1). Blood results were initially disbelieved as the patient was so well, but the results were rapidly checked and returned as similar.
Table 10.1
Blood results from patients with a long QT
Patient value | Normal range | |
|---|---|---|
Sodium | 140 | (133–146) |
Potassium | 3.8 | (3.5–5.3) |
Urea | 35.2 | (2.5–7.8) |
Creatinine | 924 | (60–120) |
Corrected Calcium | 1.09 | (2.20–2.60) |
Inorganic Phosphate | 2.43 | (0.80–1.50) |
Magnesium | 0.55 | (0.7–1.0) |
Despite looking remarkably well, a diagnosis of renal failure was raised and the patient was urgently referred to a nephrologist. He was subsequently found to have bilateral hydronephrosis with presumed chronic urinary retention. Further investigation revealed that he suffered from undiagnosed posterior urethral valves. Immediate dialysis was started but unfortunately damage proved irreversible, and he is currently awaiting renal transplant. On further investigation it was realised that initial blood tests some 2 months beforehand had been abnormal but not acted upon. The repeated ECGs once started on dialysis showed improvement of the QT interval duration with completely normalization of the measurement.
Discussion
Congenital long QT syndrome is an important cause of cardiac death in young people. It is one of the channelopathies that can present with syncope, aborted cardiac arrest or sudden death in otherwise seemingly completely well young people.
The exact prevalence is unknown but an incidence of 1:10,000 has been proposed. The condition may be lethal and in approximately 13 % of cases sudden death is the first manifestation. As a consequence screening of first degree relatives of anyone diagnosed is strongly advised so that asymptomatic people may be identified and protected with betablockers before their first symptom.
In some countries all sportsmen and women undergo compulsory screening before they are allowed to compete. In the UK, many professional clubs in different sports now insist on screening. Screening of the general population, however, has not been adopted and remains controversial. Screening for long QT syndrome is complicated by the lability of the QT interval, the absence of a long QT in some people who have the genetic mutation, the difficulty in measuring and correcting the interval and the problem of the interpretation of borderline findings.
Congenital long QT syndrome may be inherited as it can be caused by a genetic mutation. There are 13 different types now recognised. The exact mutation can be identified in up to 75 % of cases with good genetic testing with 90 % of the cases genetically identified being long QT 1, 2 or 3. Once found, the mutation allows more specific treatment and lifestyle advice for the index case and allows genetic screening to be offered to relatives.
If the genetic mutation cannot be found, or, prior to genetic analysis, screening is performed clinically by ECG, exercise testing ±24-h tape. It is important to remember that a significant minority (25 %) of the patients with long QT syndrome confirmed by the presence of a mutation of one of the long QT genes may have a normal QTc interval on the ECG.
Some cases of long QT syndrome arise “de novo,” meaning that the mistake in the DNA has arisen for the first time in that individual and that person will be the first affected in a family. It is useful to bear in mind that when assessing a patient without a family history the pre test probability for long QT syndrome is 1 in 2000, whereas for a relative of someone with confirmed long QT syndrome the probability can be 50 %.
Making a diagnosis of long QT syndrome can be difficult and challenging even for experienced physicians. The QT interval is labile and can be challenging to measure.
The normal QT interval adapts with RR interval (rate). To account for this a mathematical formula has been adopted – Bazetts’ formula. Although known not to totally adjust at higher and lower heart rates it has been universally accepted and used by physicians worldwide. Whilst the upper limit of the QTc interval is often quoted as 440 in males and 460 in females the recent guidelines for diagnosis for long QT syndrome recommend at least two ECGs with a corrected QT interval of 480 or greater. Not only is the duration of the QT interval important but also the morphology of the T wave. It has been reported that there are different shapes of T wave that are characteristic of the different types of long QT. A broad-based T wave may be seen in long QT 1, whereas T waves are typically low amplitude or bifid in long QT2, and in the long QT3 late-onset T waves are characteristic. In retrospect our patient had very tall peaked T waves characteristic of a metabolic abnormality.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


