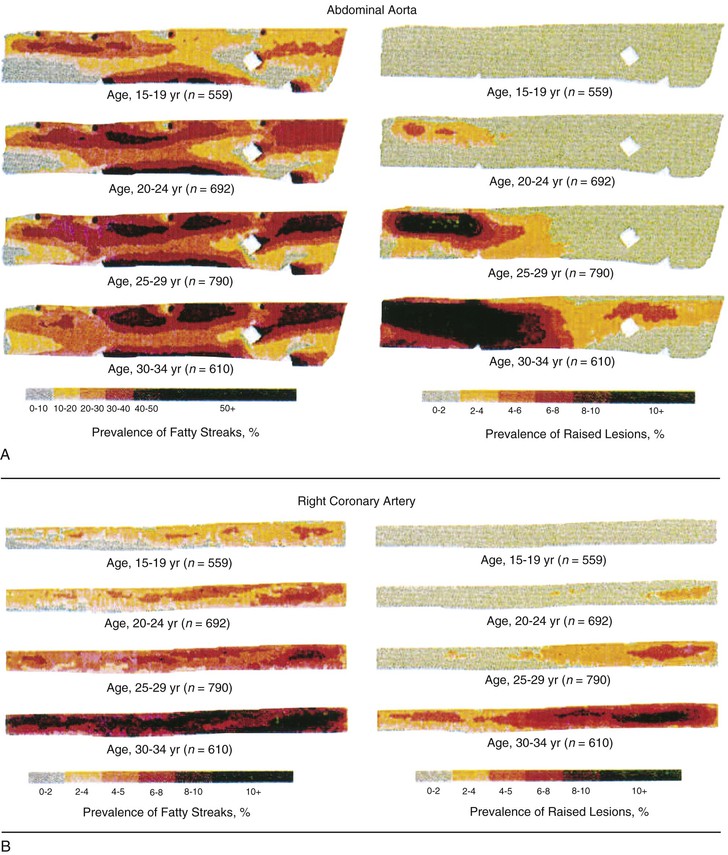
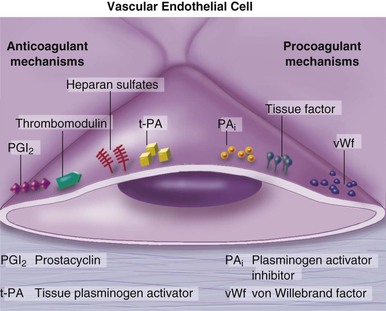
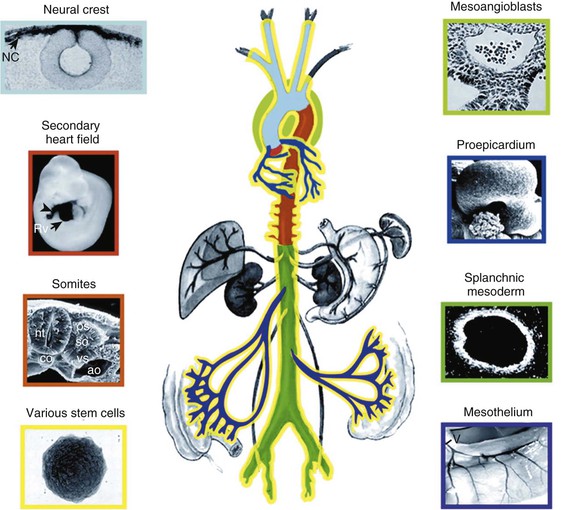
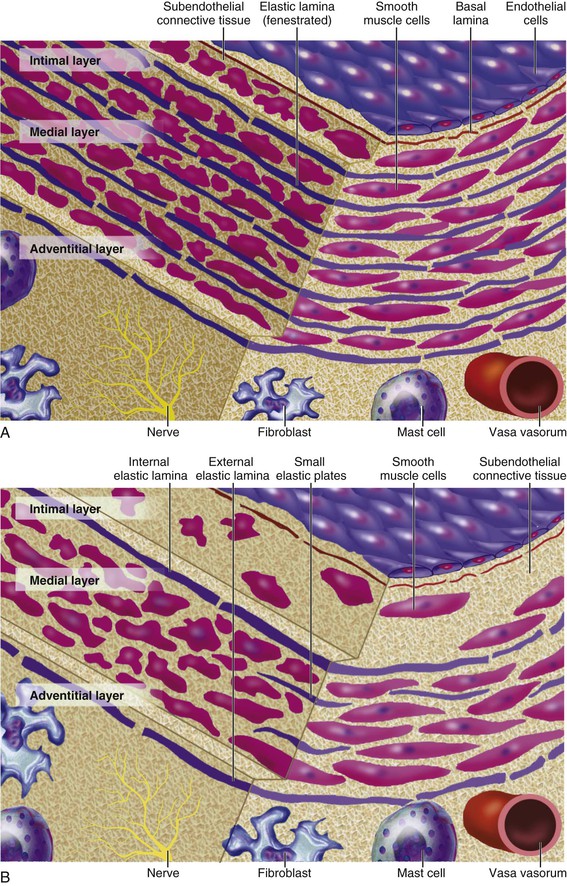
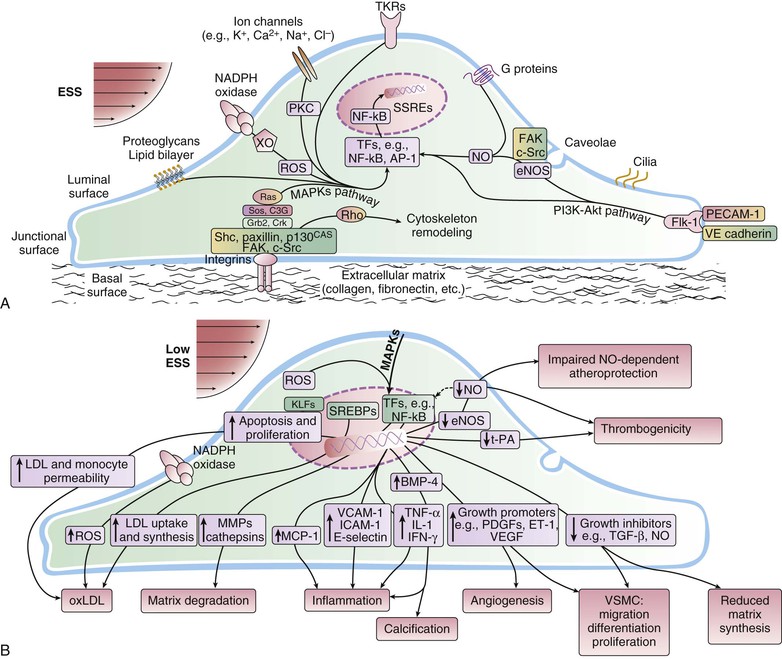
Peter Libby The 20th century witnessed a remarkable evolution in concepts concerning the pathogenesis of atherosclerosis. This disease has a venerable history, having left traces in the arteries of Egyptian mummies.1 Atherosclerosis became epidemic as populations increasingly survived early mortality associated with communicable diseases and malnutrition. Economic development and urbanization promoted habits of poor diet (e.g., a surfeit of saturated fats) and diminished physical activity, which can favor atherogenesis (see Chapters 1, 42, and 45). These environmental factors have spread steadily, such that we face an epidemic of atherosclerosis that reaches far beyond Western societies. Today, arteries are no longer viewed as inanimate tubes. In the mid-19th century, Rudolf Virchow recognized the participation of cells in atherogenesis. A controversy raged between Virchow, who viewed atherosclerosis as a proliferative disease, and Carl von Rokitansky, who believed that atheroma derived from healing and resorption of thrombi.2 Experiments performed in the early part of the 20th century used dietary modulation to produce fatty lesions in the arteries of rabbits and ultimately identified cholesterol as the culprit.3 These observations, followed by the characterization of human lipoprotein particles in the mid-20th century, promoted the insudation of lipids as a cause for atherosclerosis. Elements of all these mechanisms indeed contribute to atherogenesis. This chapter summarizes evidence from human studies, animal experimentation, and in vitro work and presents a synoptic view of atherogenesis from the biologic perspective. Acquaintance with the vascular biology of atherosclerosis should prove useful to the practitioner. Our daily contact with this common disease lulls us into a complacent belief that we understand it better than we actually do. For example, we have begun to understand why atherosclerosis affects certain regions of the arterial tree preferentially and why its clinical manifestations occur only at certain times. Atherosclerosis can involve both large and mid-size arteries diffusely. Postmortem and intravascular ultrasound clinical studies have revealed widespread intimal thickening in patients with atherosclerosis. Many asymptomatic persons have intimal lesions in their coronary or carotid arteries even in the early decades of life. At the same time, atherosclerosis produces focal stenoses in certain areas of affected vessels much more often than in others. The biologic basis of the predilection of certain sites to develop atherosclerotic lesions has begun to emerge. Atherosclerosis also displays heterogeneity in time; this disease has both chronic and acute manifestations. Few human diseases have a longer “incubation” period than atherosclerosis, which begins to affect the arteries of many Americans in the second and third decades of life (Fig. 41-1). Indeed, many young Americans have abnormal thickening of the coronary arterial intima; yet typically, symptoms of atherosclerosis emerge only after several decades of delay, characteristically appearing even later in women. Despite this indolent time course and prolonged period of clinical inactivity, the dreaded complications of atheroma—such as myocardial infarction, unstable angina, and stroke—typically occur suddenly and often without warning. Another poorly understood issue regarding atherogenesis is its role in the narrowing, or stenosis, of some vessels and in the dilation, or ectasia, of others. Traditionally, cardiologists have focused on stenoses in coronary arteries, but atherosclerosis commonly manifests as aneurysms—for example, in the aorta. Even in the life history of a single atherosclerotic lesion, a phase of ectasia known as positive remodeling, or compensatory enlargement, precedes the formation of stenotic lesions. Contemporary vascular biology has begun to shed light on some of these puzzling aspects of atherosclerosis. The endothelial cell (EC) of the arterial intima constitutes the crucial contact surface with blood. Arterial ECs possess many highly regulated mechanisms of capital importance in vascular homeostasis that often go awry during the pathogenesis of arterial diseases. For example, the EC provides one of the only surfaces, either natural or synthetic, that can maintain blood in a liquid state during protracted contact (Fig. 41-2). This remarkable blood compatibility derives in part from the expression of heparan sulfate proteoglycan molecules on the surface of the EC. These molecules, like heparin, can serve as a cofactor for antithrombin III, causing a conformational change that allows this inhibitor to bind to and inactivate thrombin. The surface of the EC also contains thrombomodulin, which binds thrombin molecules and can exert antithrombotic properties by activating proteins S and C. Should a thrombus begin to form, the normal EC possesses potent fibrinolytic mechanisms associated with its surface. The EC can produce both tissue- and urokinase-type plasminogen activators. These enzymes—t-PA and u-PA, respectively—catalyze the activation of plasminogen to form plasmin, a fibrinolytic enzyme. (For a complete discussion of the role of endothelium in hemostasis and fibrinolysis, see Chapter 82.) ECs have a common origin but acquire “bed-specific” characteristics during development. The ECs that form the inner lining of all blood vessels arise during embryogenesis from regions known as the blood islands, located on the embryo’s periphery. Angioblasts, the predecessors of ECs, share this site with the precursors of blood cells. Despite arising from the same site, cells display considerable heterogeneity even during embryologic and early postnatal development. Although ECs presumably derive from a common precursor, the signals they encounter during vessel development differ. As rudimentary blood vessels begin to form, endothelial precursors interact with surrounding cells. The interchange permits spatial and temporal gradients of various stimuli and their receptors on the ECs, leading to this cell type’s heterogeneity in the adult. EC heterogeneity depends on both environmental stimuli and epigenetic features acquired during development.4–6 Cells that make up various compartments of the arterial wall may originate from bone marrow during postnatal life as well as from their traditional embryologic sources. In particular, peripheral blood appears to contain endothelial precursor cells that may help repair areas of endothelial desquamation.7 Some experimental evidence has challenged the notion that endothelial progenitor cells (EPCs) populate murine atherosclerotic plaques.8 The second major cell type of the normal artery wall, the smooth muscle cell (SMC), has many important functions in normal vascular homeostasis, as a target of therapies in cardiovascular medicine, and in the pathogenesis of arterial diseases. These cells contract and relax and thus control blood flow through the various arterial beds, generally at the level of the muscular arterioles. In the larger types of arteries involved in atherosclerosis, however, abnormal smooth muscle contraction may cause vasospasm, a complication of atherosclerosis that may aggravate the embarrassment of blood flow. SMCs synthesize the bulk of the complex arterial extracellular matrix that plays a key role in normal vascular homeostasis and in the formation and complication of atherosclerotic lesions. These cells also can migrate and proliferate, contributing to the formation of intimal hyperplastic lesions, including atherosclerosis and restenosis; stent stenosis after percutaneous intervention; or anastomotic hyperplasia, complicating vein grafts. Death of SMCs may promote destabilization of atheromatous plaques or may favor ectatic remodeling and ultimately aneurysm formation. In contrast with ECs, thought to derive from a common precursor, SMCs can arise from many sources9 (Fig. 41-3). After ECs form tubes, the rudimentary precursor of blood vessels, they recruit the cells that will become SMCs or pericytes (smooth muscle–like cells associated with microvessels). In the descending aorta and arteries of the lower body, the regional mesoderm serves as the source of smooth muscle precursors. The mesodermal cells in somites give rise to the SMCs that invest much of the distal aorta and its branches. In arteries of the upper body, however, SMCs actually can derive from a completely different germ layer—neurectoderm, rather than mesoderm. Before the neural tube closes, neuroectodermal cells migrate and become the precursors of SMCs in the ascending aorta and some of its branches, including the carotid arteries. SMCs in the coronary arteries derive from mesoderm, but in a special way: The precursors of coronary artery SMCs arise from yet another embryologic source, a structure known as the proepicardial organ. Lineage analyses indicate that large patches of SMCs in arteries arise as expansions of small clones established early in development.9 A small population of precursor cells may reside in the tunica media of normal arteries that give rise to the SMCs that accumulate in injured or atherosclerotic arteries.10,11 The heterogeneity of SMCs may have direct clinical implications for explicating several common observations, such as the propensity of certain arteries or regions of arteries to develop atherosclerosis or heightened responses to injury (e.g., the proximal left anterior descending coronary artery), and medial degeneration (e.g., the proximal aorta in Marfan syndrome). Differential responses of SMCs to regulators of extracellular matrix production help explain why the clinical manifestations of systemic defects in fibrillin and elastin characteristically occur locally in the ascending aorta (see Chapter 57).12 The plasticity of SMCs may even extend to giving rise to cells with characteristic and functions of mononuclear phagocytes in atherosclerotic plaques.13 An understanding of the pathogenesis of atherosclerosis first requires knowledge of the structure and biology of the normal artery and its indigenous cell types. Normal arteries have a well-developed trilaminar structure (Fig. 41-4). The innermost layer, the tunica intima, is thin at birth in humans and in many nonhuman species. Although it often is depicted as a monolayer of ECs abutting directly on a basal lamina, the structure of the adult human intima is actually much more complex and heterogeneous. The endothelial monolayer resides on a basement membrane containing nonfibrillar collagen types, such as type IV collagen, laminin, fibronectin, and other extracellular matrix molecules. With aging, human arteries develop a more complex intima containing arterial SMCs and fibrillar forms of interstitial collagen (types I and III). SMCs produce these extracellular matrix constituents of the arterial intima. The presence of a more complex intima, known by pathologists as diffuse intimal thickening, characterizes most adult human arteries. Some locales in the arterial tree tend to develop a thicker intima than other regions, even in the absence of atherosclerosis (Fig. 41-5). For example, the proximal left anterior descending coronary artery often contains a more fully developed intimal cushion of SMCs than that in typical arteries. The diffuse intimal thickening process does not necessarily go hand in hand with lipid accumulation and may occur in persons without substantial burden of atheroma. The internal elastic membrane bounds the tunica intima abluminally and serves as the border between the intimal layer and the underlying tunica media. The tunica media lies under the intima and internal elastic lamina. The media of elastic arteries such as the aorta has well-developed concentric layers of SMCs, interleaved with layers of elastin-rich extracellular matrix (see Fig. 41-4A). This structure appears well adapted to the storage of the kinetic energy of left ventricular systole by the walls of great arteries. The lamellar structure also certainly contributes to the structural integrity of the arterial trunks. The media of smaller muscular arteries usually has a less stereotyped organization (see Fig. 41-4B). SMCs in these smaller arteries generally embed in the surrounding matrix in a more continuous than lamellar array. The SMCs in normal arteries seldom proliferate. Indeed, rates of both cell division and cell death are low under usual circumstances. In the normal artery, a state of homeostasis of extracellular matrix also typically prevails. Because extracellular matrix neither accumulates nor atrophies, rates of arterial matrix synthesis and dissolution usually balance each other. The external elastic lamina bounds the tunica media abluminally, forming the border with the adventitial layer. The adventitia of arteries typically has received little attention, although appreciation of its potential roles in arterial homeostasis and pathology has increased. The adventitia contains collagen fibrils in a looser array than that usually encountered in the intima. Vasa vasorum and nerve endings localize in this outermost layer of the arterial wall. The cellular population in the adventitia is sparser than in other arterial layers. Cells encountered in this layer include fibroblasts and mast cells (see Fig. 41-4). Emerging evidence suggests a role for mast cells in atheroma and aneurysm formation in animal models, but their importance in humans remains speculative.14,15 The first steps in human atherogenesis remain largely conjectural, but the integration of observations of tissues obtained from young humans with the results of experimental studies of atherogenesis in animals provides hints in this regard. On initiation of an atherogenic diet, typically rich in cholesterol and saturated fat, small lipoprotein particles accumulate in the intima (Fig. 41-6, steps 1 and 2). These lipoprotein particles appear to decorate the proteoglycan of the arterial intima and tend to coalesce into aggregates (Fig. 41-7). Detailed kinetic studies of labeled lipoprotein particles indicate that a prolonged residence time characterizes sites of early lesion formation in rabbits. The binding of lipoproteins to proteoglycan in the intima leads to their capture and retention, accounting for their prolonged residence time. Lipoprotein particles bound to proteoglycan have increased susceptibility to oxidative or other chemical modifications, considered by many to contribute to the pathogenesis of early atherosclerosis (step 2 in Fig. 41-6,). Other studies suggest that permeability of the endothelial monolayer increases at sites of lesion predilection to low-density lipoprotein (LDL). Contributors to oxidative stress in the nascent atheroma could include reduced nicotinamide adenine dinucleotide/nicotinamide adenine dinucleotide phosphate (NADH/NADPH) oxidases expressed by vascular cells, lipoxygenases expressed by infiltrating leukocytes, or the enzyme myeloperoxidase. Another hallmark of atherogenesis, leukocyte recruitment and accumulation (step 4 in Fig. 41-6), also occurs early in lesion generation (Fig. 41-8). The normal EC generally resists adhesive interactions with leukocytes. Even in inflamed tissues, most recruitment and trafficking of leukocytes occurs in postcapillary venules and not in arteries. Very soon after initiation of hypercholesterolemia, however, leukocytes adhere to the endothelium and move between EC junctions, or even penetrate through ECs (transcytosis) to enter the intima, where they begin to accumulate lipids and become foam cells16 (step 5 in Fig. 41-6) (see Fig. 41-8). In addition to the monocyte, T lymphocytes also tend to accumulate in early human and animal atherosclerotic lesions. The expression of certain leukocyte adhesion molecules on the surface of the EC regulates the adherence of monocytes and T cells to the endothelium.17 Several categories of leukocyte adhesion molecules exist. Members of the immunoglobulin superfamily include structures such as vascular cell adhesion molecule 1 (VCAM-1), or CD106. This adhesion molecule is of particular interest in the context of early atherogenesis because it interacts with an integrin (very late antigen 4 [VLA-4]) characteristically expressed by only those classes of leukocytes that accumulate in nascent atheroma—monocytes and T cells. Moreover, experimental studies have shown expression of VCAM-1 on ECs overlying very early atheromatous lesions. Other members of the immunoglobulin superfamily of leukocyte adhesion molecules include intercellular adhesion molecule 1 (ICAM-1). This molecule is more promiscuous, both in the types of leukocytes it binds and in its wide and constitutive expression at low levels by ECs in many parts of the circulation. Selectins constitute the other broad category of leukocyte adhesion molecules. The prototypical selectin, E-selectin or CD62E (E stands for “endothelial,” the cell type that selectively expresses this particular family member), probably has little to do with early atherogenesis. E-selectin preferentially recruits polymorphonuclear leukocytes, a cell type seldom found in early atheromata (but an essential protagonist in acute inflammation and host defenses against bacterial pathogens). Moreover, ECs overlying atheroma do not express high levels of this adhesion molecule. Other members of this family, including P-selectin, or CD62P (P stands for “platelet,” the original source of this adhesion molecule), may play a greater role in leukocyte recruitment in atheroma, because ECs overlying human atheroma express this adhesion molecule. Selectins tend to promote saltatory or rolling locomotion of leukocytes over the endothelium. Adhesion molecules belonging to the immunoglobulin superfamily tend to promote tighter adhesive interactions and immobilization of leukocytes. Studies in genetically altered mice have proven roles for VCAM-1 and P-selectin (including both platelet-derived and endothelium-derived P-selectin) in experimental atherosclerosis. Increasing evidence supports the accumulation in atheromas of distinct subtypes of mononuclear phagocytes.16,18–21 The functional consequences of this heterogeneity of macrophage populations in plaques require further study, especially in humans. In mice, a particularly proinflammatory subset of monocytes accumulates in the spleen and peripheral blood in response to hypercholesterolemia and preferentially populates nascent atheroma.21 Once adherent to the endothelium, leukocytes must receive a signal to penetrate the endothelial monolayer and enter the arterial wall (step 4 in Fig. 41-6). The current concept of directed migration of leukocytes involves the action of protein molecules known as chemoattractant cytokines or chemokines.16,22 Among the many chemokines implicated in atherogenesis, two are of particular interest in recruiting the mononuclear cells characteristic of the early atheroma. One such molecule, known as monocyte chemoattractant protein 1 (MCP-1), or CCL2, is produced by the endothelium in response to oxidized lipoprotein and other stimuli. Cells intrinsic to the normal artery, including ECs and SMCs, can produce this chemokine when stimulated by inflammatory mediators, as do many other cell types. MCP-1 selectively promotes the directed migration, or chemotaxis, of monocytes. Atherosclerosis-prone mice lacking MCP-1 or its receptor CCR2 exhibit delayed and attenuated lesion formation. Human atherosclerotic lesions express increased levels of MCP-1 compared with uninvolved vessels. In mice, the CCL2/CCR2 dyad recruits preferentially the proinflammatory monocyte subset.21 Fractalkine, a unique cell surface–bound chemokine, also appears to contribute to atherogenesis and typically interacts with the less inflammatory subset of monocytes.23 Another group of chemoattractant cytokines may heighten lymphocyte accumulation in plaques as well: Atheromas express a trio of lymphocyte-selective chemokines (IP-10 or CXCL10, I-TAC or CXCL11, and MIG or CXCL9). Interferon-γ, a cytokine known to be present in atheromatous plaques, induces the genes encoding this family of T cell chemoattractants. The accumulation of monocytes in plaques depends not only on their recruitment, but also on their retention.24 Recent work has implicated netrin-1 interacting with its receptor UNC5b (both induced by hypoxia) as a protein that retards macrophages from exiting plaques.25–27 The spatial heterogeneity of atherosclerosis is challenging to explain in mechanistic terms. Equal concentrations of bloodborne risk factors such as lipoproteins bathe the endothelium throughout the vasculature. It is difficult to envisage how injury due to inhalation of cigarette smoke could produce any local rather than global effect on arteries, yet stenoses due to atheromas typically form focally. Some researchers have invoked a multicentric origin hypothesis of atherogenesis, positing that atheromas arise as benign leiomyomas of the artery wall. The monotypia of various molecular markers in individual atheromas supports this monoclonal hypothesis of atherogenesis.9 The location of sites of lesion predilection at proximal portions of arteries after branch points or bifurcations at flow dividers, however, suggests a hydrodynamic basis for early lesion development. Arteries without many branches (e.g., the internal mammary and radial arteries) tend not to develop atherosclerosis. Two concepts can aid in understanding how local flow disturbances might render certain foci sites of lesion predilection. Locally disturbed flow could induce alterations that promote the steps of early atherogenesis. Alternatively, the laminar flow that usually prevails at sites that do not tend to develop early lesions may elicit antiatherogenic homeostatic mechanisms (atheroprotective functions).28 The EC experiences the laminar shear stress of normal flow and the disturbed flow (usually yielding decreased shear stress) at sites of predilection.29 Multiple mechanotransduction mechanisms operate to signal the local shear stress environment to ECs. For example, these cells have cilia on their luminal surface and adhesion receptors in their lateral cell membrane that can sense tension, transmit forces to the cortical cytoskeleton, and potentially regulate ion channels or G protein–coupled receptors that signal changes in gene expression (Fig. 41-9A).29,30 In vitro data suggest that laminar shear stress can augment the expression of genes that may protect against atherosclerosis, including forms of the enzymes superoxide dismutase and nitric oxide synthase.28 Superoxide dismutase can reduce oxidative stress by catabolizing the reactive and injurious superoxide anion. Endothelial nitric oxide synthase produces the well-known endogenous vasodilator nitric oxide. Beyond its vasodilating actions, however, nitric oxide can resist inflammatory activation of endothelial functions, such as expression of the adhesion molecule VCAM-1. Nitric oxide appears to exert this anti-inflammatory action at the level of gene expression by interfering with the transcriptional regulator nuclear factor κB (NF-κB). Nitric oxide increases the production of IκBα, an intracellular inhibitor of this important transcription factor. NF-κB regulates numerous genes involved in inflammatory responses in general, and in atherogenesis in particular.
The Vascular Biology of Atherosclerosis
Overview and Background
Structure of the Normal Artery
Cell Types Composing the Normal Artery
Endothelial Cells
Arterial Smooth Muscle Cells
Layers of the Normal Artery
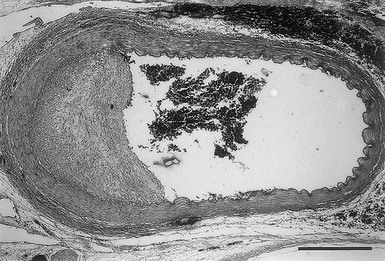
Intima
Tunica Media
Adventitia
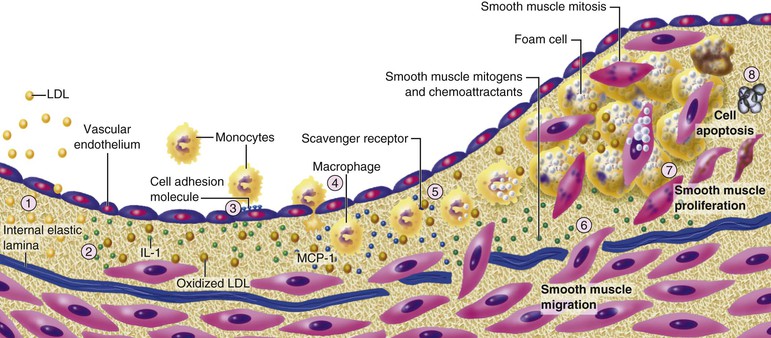
Atherosclerosis Initiation
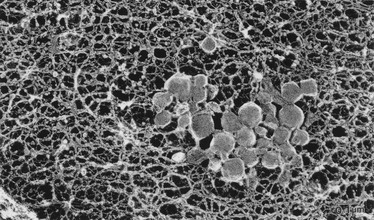
Extracellular Lipid Accumulation
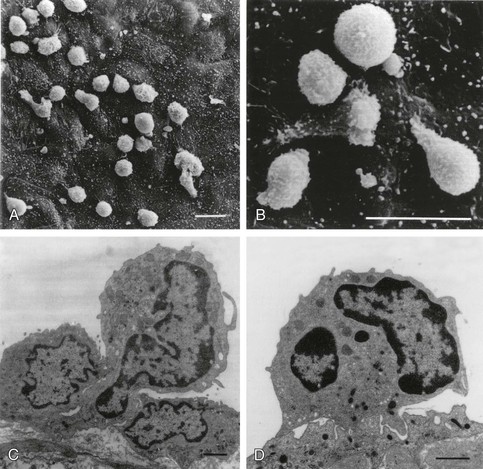
Leukocyte Recruitment and Retention
Focality of Lesion Formation