8 OBJECTIVES
Pulmonary Vascular Disease
![]() Identify the components of the pulmonary vasculature and discuss the pathophysiology and causes of hypoxemia and its relation to pulmonary hypertension.
Identify the components of the pulmonary vasculature and discuss the pathophysiology and causes of hypoxemia and its relation to pulmonary hypertension.
![]() Evaluate the other causes of pulmonary hypertension.
Evaluate the other causes of pulmonary hypertension.
![]() Review the clinical implications and management of venous thromboembolic disease and pulmonary embolism.
Review the clinical implications and management of venous thromboembolic disease and pulmonary embolism.
INTRODUCTION
Unlike most of the other organs in the body, the lung has dual circulation. It receives blood flow from both the pulmonary and systemic circulations. The pulmonary arteries and arterioles carry mixed-venous blood from the right ventricle and distribute it over the pulmonary capillary-alveolar membrane, where gas exchange takes place. Oxygenated blood is then returned to the left atrium via the pulmonary venules and veins. The nutrient requirements of respiratory bronchioles and the elastic components of the lung are met by the pulmonary circulation, while their oxygen requirements are principally satisfied directly by alveolar ventilation. In contrast, the bronchial circulation arises from the aorta and supplies the tracheobronchial tree with oxygen and nutrients to the level of the terminal bronchioles. The pulmonary circulation is normally a high-compliance, low-resistance, low-pressure system that receives the entire output of the right ventricle, approximately 3.5 L/min/M2. In contrast, the bronchial circulation is a low-compliance, high-resistance system with a blood flow of approximately 70 mL/min/M2 and a pressure equal to aortic pressure. The bronchial circulation can have important clinical implications as discussed later in this chapter.
In health, the pulmonary vascular bed is a thin-walled, high-compliance, low-resistance system with a resting mean pulmonary artery pressure of approximately 12–14 mm Hg, a pulmonary capillary pressure of 6–8 mm Hg, and a pulmonary venous pressure of 5–7 mm Hg. During conditions in which pulmonary blood flow is transiently increased, such as exercise, the ability to recruit and distend pulmonary vessels permits up to a sevenfold increase in cardiac output with less than a doubling of mean pulmonary artery pressure. Elevation of hydrostatic pressures beyond this normal range will produce distinct clinical syndromes depending on whether the pressure elevation is precapillary (arteries and arterioles) or postcapillary (venules, veins, or left side of the heart).
The primary determinants of mean pulmonary artery pressure under normal circumstances are left atrial pressure, pulmonary blood flow, and pulmonary vascular resistance. The clinical causes related to these determinates are outlined (Table 8–1).
Table 8–1. Pathophysiological classification & causes of pulmonary hypertension
I. Diseases associated with chronic increases in pulmonary blood flow |
A. Congenital left-to-right shunts |
1. Intracardiac |
a. Atrial septal defects |
b. Ventricular septal defects |
2. Extracardiac |
a. Patent ductus arteriosus |
II. Diseases that increase pulmonary vascular resistance |
A. Increased resistance primarily in precapillary arteries and arterioles |
1. Increased resistance caused by hypoxic vasoconstriction |
a. Cause of hypoxemia low inspired partial pressure of oxygen |
i. Chronic residence at high altitude |
b. Cause of hypoxemia chronic or recurrent alveolar hypoventilation |
i. Morbid obesity |
ii. Neuromuscular weakness |
iii. Severe kyphoscoliosis |
iv. Obstructive sleep apnea |
c. Cause of hypoxemia primarily ventilation-perfusion mismatch |
i. Chronic obstructive pulmonary disease |
ii. Chronic bronchitis |
iii. Asthma |
iv. Emphysema |
v. Bronchiectasis |
vi. Chronic interstitial lung diseases |
2. Increase in resistance caused by intraluminal obstruction, fibrosis, or destruction of arteries or arterioles |
a. Pulmonary embolism |
b. Schistosomiasis |
c. Sickle cell disease |
d. Pulmonary fibrosis |
e. Pulmonary vasculitis |
f. Toxins |
i. Anoretic drugs |
ii. Cocaine |
iii. Intravenous drugs |
g. Human immunodeficiency virus (HIV) |
h. Cirrhosis with portal hypertension |
i. Primary pulmonary hypertension |
B. Increase in resistance primarily in postcapillary venules and veins |
1. Pulmonary venoocclusive disease |
2. Sclerosing mediastinitis |
3. Anomalous venous return |
III. Diseases that increase left atrial pressure |
A. Left ventricular systolic dysfunction |
B. Left ventricular diastolic dysfunction |
1. Hypertrophic cardiomyopathy |
2. Constrictive pericarditis |
C. Valvular heart disease |
1. Mitral stenosis |
2. Mitral regurgitation |
D. Miscellaneous |
1. Left atrial myxoma |
PRECAPILLARY PULMONARY HYPERTENSION
Pathophysiology
Pulmonary arterial hypertension (PAH) is the term used to describe the remodeling of pulmonary vessels in a fashion that leads to an increase in vascular resistance and pulmonary artery pressure. For epidemiologic purposes, pulmonary hyper-tension is defined as a mean pulmonary artery pressure greater than 25 mm Hg at rest or greater than 30 mm Hg during exercise. Both genetic and epigenetic risk factors for PAH have been identified in patients with PAH.
A majority of patients with familial PAH have mutations in the bone morphogenic protein receptor-2 (BMPR2) gene that codes for type II receptors in the TGF-β superfamily. These mutations increase the propensity for unregulated growth and proliferation of smooth muscle cells and fibroblasts in vessel walls. Many of these mutations are inherited in a Mendelian dominant fashion and demonstrate incomplete penetrance and genetic anticipation. Incomplete penetrance means that not all individuals with the mutation will be affected. Genetic anticipation means that subsequent generations often develop the symptoms of the disease at an earlier age than previous generations.
In contrast to gene mutations, epigenetic factors represent injuries, insults, or pathophysiologic states that can influence the expression of even normal genes in a way that can lead to disease. In the case of PAH, these insults can initiate the process of vascular remodeling that leads to pulmonary hypertension. Some of these epigenetic risk factors are well known, such as congenital left-to-right shunts, connective tissue diseases, use of selected anorectic drugs, portal hypertension, and infection with the human immunodeficiency virus (HIV).
Regardless of the etiology of PAH, the endothelium from pulmonary arteries of patients with PAH produces fewer naturally occurring vasodilators and growth inhibitors such as prostacyclin and nitric oxide, and more vasoconstrictors and growth stimulators such as thromboxane and endothelin-1. This imbalance in the controllers of vascular tone and vascular remodeling in the lung is thought to be an important pathogenic mechanism in the development of PAH.
Atrial septal defects, ventricular septal defects, patent ductus arteriosus, and other congenital heart diseases associated with shunting of blood from the systemic to the pulmonary circulation can create a condition of chronic overcirculation and overdistension of the pulmonary vascular bed. When significant left-to-right shunts are not diagnosed and corrected in early childhood, the resultant chronic vascular wall stress induces the process of remodeling. Remodeling is characterized histologically by medial hypertrophy, intimal thickening, and adventitial fibrosis of pulmonary arteries and arterioles. These changes further increase pulmonary vascular resistance and eventually become irreversible. In such cases, pulmonary hypertension may continue to worsen after shunt correction. Late in the disease process, right-sided pressures can exceed systemic pressures, causing a reversal of the shunt. In such cases, termed Eisenmenger’s complex, poorly oxygenated mixed-venous blood bypasses the lungs and enters the systemic circulation, causing severe hypoxemia.
Fibrosing lung diseases may also cause precapillary pulmonary hypertension by destroying small arterioles and alveolar capillaries. When the destruction is severe enough and the cross-sectional area of the pulmonary vascular bed is significantly reduced, pulmonary vascular resistance rises. Although connective tissue diseases can cause pulmonary fibrosis, they often cause pulmonary hypertension that is out of proportion to the severity of either the fibrosis or the resultant hypoxemia. This is particularly characteristic of limited scleroderma, a disease in which up to 40%–90% of patients will develop PAH. In scleroderma, deposition of collagen in the walls of small arterioles occurs in the lung as well as in other organs, contributing to the increase in pulmonary vascular resistance.
Hypoxemia is the term used to describe a decrease in the partial pressure of oxygen in the systemic arterial blood. It can contribute to the development of pre-capillary pulmonary hypertension by inducing hypoxic vasoconstriction in pulmonary arterioles. Hypoxemia is one of the most common manifestations of cardiopulmonary disease and results from one of six pathophysiologic mechanisms:
1. decreased atmospheric pressure
2. decreased inspired fraction of oxygen
3. alveolar hypoventilation
4. ventilation-perfusion mismatch
5. shunt
6. diffusion abnormalities
In the United States the most common disease associated with hypoxemia is chronic obstructive pulmonary disease (COPD). In patients with COPD, the severity of pulmonary hypertension is generally modest with mean pulmonary artery pressures rarely exceeding 30 mm Hg. Chronic alveolar hypoventilation, also often seen in COPD, can exaggerate the hypoxic vasoconstrictor response. Pulmonary hypertension occurs in association with other causes of significant hypoxemia and alveolar hypoventilation, such as morbid obesity with obstructive sleep apnea or kyphoscoliosis (Table 8–1). In hypoxemic patients, enhanced erythropoietin production can raise the blood hematocrit and viscosity, further aggravating the pulmonary hypertension and increased right ventricular workload. When hypoxemia is chronic, vascular remodeling occurs, leading to persistent pulmonary hypertension. Supplemental oxygen in this setting may not completely resolve the pulmonary hypertension.
Pulmonary embolism is an important and reversible cause of precapillary pulmonary hypertension. Venous thromboembolism is a term used to describe the spectrum of clinical presentations of deep vein thrombosis and pulmonary embolism. Venous thromboembolism remains an important cause of mortality in the United States. It has been estimated that there are 5 million cases of deep vein thrombosis that result in 500,000 cases of symptomatic pulmonary embolism each year. Approximately 10% of cases of pulmonary embolism are fatal, accounting for up to 35% of all hospital deaths. It has been estimated that over 70% of patients who die of pulmonary embolism are diagnosed with pulmonary embolism for the first time at autopsy. Late complications of venous thromboembolism include 500,000 new cases of venous stasis ulcers each year in the United States. This diagnosis will be dealt within more detail later in the chapter.
Some diseases do not fit well into any of the above categories, such as hemoglobin SS, schistosomiasis, and sarcoidosis. The pathogenesis of pulmonary hypertension in these diseases is variable. For example, in schistosomiasis there is obstruction of pulmonary vessels both from the schistosomes themselves and from the intense perivascular inflammation that accompanies the parasitic infestation, while in sarcoidosis, noncaseating granulomatous inflammation may be present.
Diagnostic & Clinical Considerations
HISTORY
 The most common presenting symptoms in patients with precapillary pulmonary hypertension are chronic dyspnea on exertion, fatigue, angina pectoris (precordial pressure-like chest pain), and occasionally syncope. Hoarseness due to compression of the recurrent laryngeal nerve by an enlarged main pulmonary artery is an uncommon but unique manifestation of pulmonary hypertension not seen in other cardiopulmonary diseases. Symptoms of hypoxemia depend on its severity and range from headache, confusion, and somnolence to clouding of the consciousness. Cardiovascular decompensation can occur with brady- or tachyarrhythmias (see Chapter 13 on respiratory failure).
The most common presenting symptoms in patients with precapillary pulmonary hypertension are chronic dyspnea on exertion, fatigue, angina pectoris (precordial pressure-like chest pain), and occasionally syncope. Hoarseness due to compression of the recurrent laryngeal nerve by an enlarged main pulmonary artery is an uncommon but unique manifestation of pulmonary hypertension not seen in other cardiopulmonary diseases. Symptoms of hypoxemia depend on its severity and range from headache, confusion, and somnolence to clouding of the consciousness. Cardiovascular decompensation can occur with brady- or tachyarrhythmias (see Chapter 13 on respiratory failure).
PHYSICAL EXAMINATION
The initial adaptive response to a chronic elevation in pulmonary artery pressure is right ventricular hypertrophy, which can reduce diastolic compliance of the right ventricle. Chronic pressure overload eventually results in reduced systolic performance as well. When cardiac output begins to fall, maladaptive compensatory mechanisms increase sodium and water reabsorption under the influences of aldosterone and antidiuretic hormone. Sodium and water retention results in volume overload, which is manifested as distended neck veins, ascites, and leg edema. This syndrome of right ventricular failure is termed cor pulmonale when the pulmonary hypertension results from lung disease or hypoxemia. If the right atrium contracts against a poorly compliant right ventricle, intraatrial pressure will rise sharply, producing a prominent “a” wave visible in the jugular veins at the base of the neck. Palpation of the precordium may reveal a parasternal lift, which is caused by contraction of the hypertrophied, dilated right ventricle just below the sternum. On auscultation, the pulmonic component of the second heart sound (P2) is often accentuated and split from the aortic component during both inspiration and expiration. Persistent splitting of the second heart sound is due to slower right ventricular ejection against increased load that results in delayed closure of the pulmonic valve. Dilation of the right ventricle and pressure overload may cause the tricuspid valve to become incompetent during systole, creating a systolic murmur heard best over the right lower sternal border. Severe tricuspid regurgitation briskly elevates central venous (CV) pressure, producing a prominent cv wave that can be visible in the jugular veins or can be palpable in the liver.
When hypoxemia is severe (partial arterial oxygen pressure [PAo2] <40 mm Hg), central cyanosis, a bluish-gray discoloration of the skin and mucous membranes, may be present. Cyanosis confined to the fingers and toes is usually indicative of poor peripheral blood flow rather than of hypoxemia. Because the symptoms and physical signs of hypoxemia are insensitive and nonspecific, arterial blood gas analysis is usually performed if the suspicion of hypoxemia is high. Pulse oxi-meters can estimate the percentage of oxygenated hemoglobin in the systemic capillary bed by comparing the transcutaneous absorption of various wavelengths of light that are specific for oxygenated and deoxygenated hemoglobin. The oxy-hemoglobin saturation estimated by a pulse oximeter may be erroneous in heavily pigmented patients or in patients with methemoglobinemia, peripheral vascular disease, or shock.
IMAGING
Chest x-rays often provide clues to the cause of pulmonary hypertension (Figure 8–1). Prominence of the main pulmonary arteries is seen in many patients with pulmonary hypertension, regardless of the cause (Figure 8–2). The contour of the cardiac silhouette may suggest either isolated right ventricular enlargement or biventricular enlargement if left ventricular dysfunction is the cause of pulmonary hypertension.
Echocardiographic studies are now used almost routinely in the evaluation of suspected or proven pulmonary hypertension, since they provide both diagnostic and prognostic information. Enlarged intracavitary dimensions, increased wall thickness, and decreased systolic function of the right ventricle often provide the first suspicion of pulmonary hypertension in patients being evaluated for dyspnea of undetermined origin (Figure 8–3). The flow velocity of the tricuspid regurgitation jet from the right ventricle into the right atrium can be measured by Doppler ultrasound technique. Because the regurgitant flow velocity increases as pulmonary artery pressure increases, this measurement can be used to estimate pulmonary artery pressure. Intracardiac shunts that cannot be visualized by standard echocardiographic techniques may be identified after the intravenous injection of micronized bubbles of air suspended in saline solution. Mixing of the echogenic bubbles with a stream of sonolucent blood can identify the site of shunting.
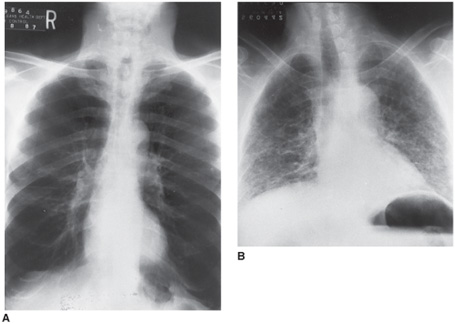
Figure 8–1. The chest x-ray in the differential diagnosis of pulmonary hypertension. A. Chest x-ray of a 60-year-old man with severe chronic obstructive pulmonary disease (COPD) due to smoking-related emphysema. Note the increased lung volume with hyper-lucent lung parenchyma. B. Chest x-ray of a 55-year-old man with idiopathic pulmonary fibrosis. The lung volumes are much smaller and the lung parenchyma has increased linear densities, which represent areas of scarring. Both patients had mean pulmonary artery pressures of approximately 30 mm Hg.
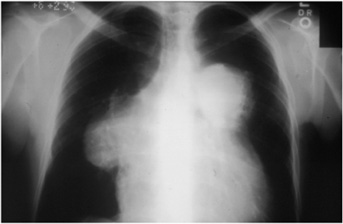
Figure 8–2. Chest x-ray showing large pulmonary arteries.
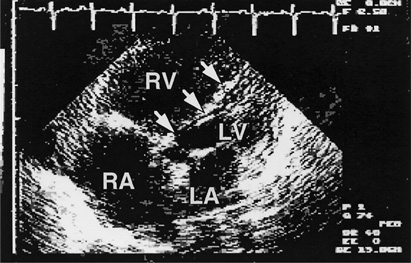
Figure 8–3. Echocardiogram in the assessment of pulmonary hypertension. This transthoracic echocardiogram from a 40-year-old woman with pulmonary hypertension due to pulmonary venoocclusive disease shows enlargement of the right atrium (RA) and right ventricle (RV) with displacement of the interventricular septum to the left (arrows), compromising filling of the left ventricle (LV). The left atrium (LA) is normal in size. Her mean pulmonary artery pressure was 60 mm Hg.
LABORATORY EVALUATION
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


