Fig. 7.1
Genetic and environmental factors in the pathogenesis of metabolic syndrome (MS)
7.2.2 Gut Microbiota Composition, Metabolic Phenotype, and Disease Risk
Several studies on animals (rats and mice) and humans demonstrate that obese and lean phenotypes are associated with different gut microbial compositions with a different Firmicutes–Bacteroidetes ratio and low levels of Methanobrevibacter smithii, the leading representative of the gut microbiota archaea [18]. Many studies showed an increase in Firmicutes and Actinobacteria in obese phenotype in animals [19] and humans [14]. These data support the Firmicutes–Bacteroidetes ratio as one of the important key factors in obesity that seems to be modulated by a weight-reduction diet.
Moreover, several research works emphasize the correlation between the concentration of certain Lactobacillus species and obesity in both humans [20] and animals [21]. Million et al. [21] observed increasing levels of Lactobacillus reuteri and depletion of Bifidobacterium animalis species in microbiota of obese test animals. Genetically, obese ob/ob mice encoding the hormone leptin promoting satiety have higher levels of Firmicutes and lower levels of Bacteroidetes at their cecal microbiota than their wild-type littermates. This condition remains unchanged when they are fed with a low-fat, polysaccharide-rich diet.
Various bacterial species were associated with obesity: Staphylococcus aureus, Escherichia coli, Faecalibacterium prausnitzii. However, other research works did not confirm the “typical change” detected by the above mentioned investigators. For example, Schwiertz et al. [18] showed an even lower Firmicutes–Bacteroidetes ratio in obese human adults compared with lean controls. They observed no differences between obese and lean subjects in the numbers of Bacteroidetes dosed in the fecal sample.
These different findings would suggest that the connection among diet, microbiota composition, and host metabolic regulation is a complex process in obesogenesis. The behavior of the microbiota–host metabolic regulation interactive axis varies in any individual according to development stages and environmental conditions such as diet, use of antibiotics and other drugs, and other factors that substantially reduce bacterial diversity [6], the contribution of the genetic background of the host being understood. Indeed, within an individual the same microbial products may achieve either a beneficial or an adverse effect. The “metacommunity theory” [22] could be used to explain the dynamics of the human microbiome, and in particular, compositional variability within and between the hosts. The “metacommunity” concept is an important way of thinking about linkages between different spatial scales in ecology. According to this theory, the diversity of the microbiome changes at different body sites, between people, with age and with diet. This leads to the realization of different environments within and between different individuals who are exposed to temporal variation and between populations. Most likely, the metagenomic-based functional aspects are more important than the difference at the phylum level of the microbiota.
7.2.3 Diet, Microbiota, and Host Metabolism
The impact of diet on microbiota composition appears to be more relevant than obesity [23]. Gut microbiota composition depends on different eating habits. In Burkina Faso children with a predominantly vegetarian diet, microbiota showed specific abundance in Bacteroidetes than Firmicutes, and bacterial augmentation of bacteria with enzymes enables hydrolysis of cellulose and xylan, and also of members of the genera Prevotella and Xylanibacter. However, Enterobacteriaceae (Shigella and Escherichia) were significantly under-represented. Otherwise, Firmicutes and Proteobacteria were more abundant in European children with a Western diet [24]. We can speculate that gut microbiota might have co-evolved with the polysaccharide-rich diet of African children, allowing them to maximize energy intake from fibers and protecting them from inflammation and non-infectious colonic diseases by modulating gut-associated lymphatic tissue (GALT). This study on microbiota composition of European and African children seems to confirm the evidence of Arumugam et al. [9] concerning three enterotypes dominated by a different genus—Bacteroides, Prevotella, or Ruminococcus.
A dietary intervention for a few days is not sufficient to change individual enterotype. A controlled feeding study of ten subjects showed that microbiome composition changed within 24 h of initiating a high-fat/low-fiber or low-fat/high-fiber diet, but that enterotype identity remained stable during the 10-day study [25].
Cani et al. [26] demonstrated in mice that a very high-fat (HF) diet induced change in gut microbiota composition associated with an increase in weight gain, adiposity, insulin resistance, and other symptoms of MS. The same authors only detected a reduction of Bifidobacterium spp. [27, 28] and Bacteroides-related bacteria, Eubacterium rectale-Blautia coccoides group, Lactobacillus spp., and Roseburia spp. content [26]. Several other studies confirmed the altered Firmicutes–Bacteroidetes ratio in mice fed with an HF diet [19, 23].
The increase in circulating bacterial endotoxins such as lipopolysaccharides (LPS) emphasizes the relationship among diet, microbiota, host metabolism, and low-grade inflammation. An HF diet induces changes in the microbiota composition accompanied by an increase in circulating Gram-negative Proteobacteria responsible, to avoid the induction of inflammatory local response and an increase in the passage of LPS from the gut lumen to the gut interstitium, where it can activate TLR4 on afferent nerve terminals [29].
Intestinal microflora change could precede the establishment of the obese phenotype and could contribute to its development. Several findings in obesity mouse models demonstrated that at a higher fat diet increases the production of Resistin-like molecule beta (RELMβ) in the stools and in the serum [30]. Furthermore, the same mechanism plays a role in humans too [30]. RELMβ is one of three isoforms of the RELM gene family [31] and it is produced by goblet cells secondary to microbial intestinal colonization [30]. The genetic activation of RELMβ is started via interleukin 4 by antigenic stimulation of the Th2 immunity response [32]. On the contrary it is inhibited by interferon γ through a Th1-mediated immunity response. RELMβ regulates the gut microbiota composition both by stimulating production of defensins, and regulating functions of the intestinal barrier at innate mucosal immunity [33], which results in macrophages activation. Moreover, a recent study proved the metabolic effects of RELMβ on intestinal homeostasis of glucose and induction of insulin resistance. RELMβ regulates intracellular transport mechanism of glucose by an increase in glucose transporter (GLUT2) expression, a passive low affinity transporter. This allows the output of glucose through the basal membranes of the enterocytes.
RELMβ expression is augmented both in genetic (ob/db mice) and nongenetic obesity murine models [34] induced by an HF diet. Indeed, an in vitro study showed how saturated fatty acids directly induce the expression of RELMβ [34].
A direct relationship has been demonstrated between expression levels of RELMβ and microbiota composition in RELMβ knockout mice; those showed an increase in Firmicutes rather than Bacteroidetes [23]. In cohort study, Hildebrandt et al. [23] demonstrated that RELMβ knockout mice remained comparatively lean when subjected to the HF diet. They found that consistent changes in microbial communities could be seen upon switching to the HF diet for both wild-type and RELMβ knockout mice. It was established that the HF diet itself, and not the obese state, was responsible for the altered microbiota. The action carried out by RELMβ on both the intestinal microbiota and the glucose homeostasis may explain the link between diet and modification of the intestinal microbiota. This could be related to the direct action carried out by RELMβ on the secretion of intestinal antimicrobial peptides. The reduced secretion of β-defensin I (and/or other members of the defensin family), induced by RELMβ, could be responsible for impaired composition of the microbiota induced by HF diets (Fig. 7.2).
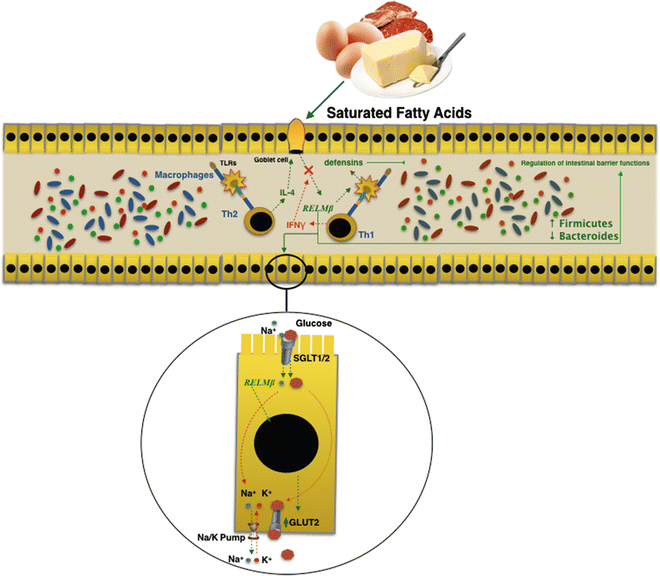
Fig. 7.2
RELMβ is produced by goblet cells in the gut in response to microbial intestinal colonization. Saturated fatty acids in a high-fat diet directly induced expression of RELMβ. The action carried out by RELMβ on both the intestinal microbiota and the glucose homeostasis may explain the link between diet and modification of the intestinal microbiota in obesity and metabolic syndrome
7.3 From Leaky Gut Syndrome and Chronic Low-Grade Inflammation to Cardiometabolic Syndrome
7.3.1 The Gut Microbiota and Innate Immunity Regulation of the Host
The mammalian immune system uses two lines of defense. Innate immunity is the first line and responds to numerous nonspecific numerous exogenous and endogenous pathogenetic factors; adaptive immunity is the second line, which gives a specific response (humoral and cell-mediated immunity) with the activation of the immune response regulated by Th1, Th2, Th17, and Th1/Th17 cells. Innate immunity represents a defensive system with an important barrier function via several mechanisms: epithelial barrier, tight junctions, antimicrobial peptides such as cathelicidins and defensins. It includes various cells: neutrophils, basophils, eosinophils, mast cells, monocytes, macrophages, dendritic cells (DCs), NK and NK-T cells, B1 cells and γδ T cells with the pathogen recognition receptors (PRRs). PRRs can be expressed on the cell surface or intracellularly. They recognize highly conserved antigens from bacteria, microbial-associated molecular patterns (MAMPs), and other pathogens, pathogen-associated molecular patterns (PAMPs). PRR activation by MAMPs, causing recruitment of down-stream signaling molecules, results in activation of NFkB transcription factor leading to increased production of pro-inflammatory cytokines [35].
Several families of innate receptors are involved in the recognition of PAMPs. They include toll-like receptors (TLRs), staying on the surface of the cellular membrane and intracellularly on a range of different cell types [36]; nucleotide oligomerization domains (NODs); inflammasomes, large intracellular cytoplasmic multi-protein complexes composed of one of several NOD-like receptors (NLRs) and pyrin and HIN domain-containing protein family (PYHIN: including NLRP1, NLRP3, NLRC4, and AIM2) playing a central role in innate immunity; C-type lectin receptors (CLRs) such as dectin-1, and RNA-sensing RIG-like helicases such as RLRs; RIG-I and MDA5; and cytosolic dsDNA sensors (CDSs). They are cytoplasmic receptors critical for host antiviral responses [37, 38] and for diverse MAMPs or death-associated molecular protein ligands released in response to cell necrosis or tissue injury: damage-associated molecular patterns (DAMPs) or alarmins.
The DAMPs contribute to “sterile inflammation” [39] via activation of TLR. Most DAMPs are substances that are normally sequestered within cells but released in response to injury. Degradation products of extracellular matrix or glycocalyx are heat shock proteins (HSP) (10), IgG chromatin complex (11), versican (12), high-mobility group box 1 (HMGB1), heparan sulfate (14), hyaluronan, uric acid, and others.
The two best characterized families of PRRs are TLRs and nucleotide oligomerization domains (NODs)-leucine-rich repeat (LRR) family (CATERPILLER family), which are intracellular receptors [37, 38]. The ligands identified for TLRs include PAMPs and DAMPs.
In the gut, TLRs and NODs are involved in maintaining mucosal and commensal homeostasis. The functional activity of the TLRs and NOD2 is very important for a healthy gut, as they are involved in host defense and tissue repair responses. Correction functioning of TLRs and NOD2 enhances barrier protection and antimicrobial activity, reduces bacterial invasion, confers tolerance, and promotes healing. When homeostasis between commensal and mucosal immunity is perturbed, TLRs and NOD2 signaling can result in an exaggerated proinflammatory responses through cytokine and chemokine production. Altered expression of TLRs induces intestinal barrier destruction, an increase in the passage of endotoxins, loss of tolerance, and antimicrobial activity with enhanced bacterial invasion [40].
Gram-negative bacteria present the LPS that binds TLR4, whereas Gram-positive bacteria have proteoglycans (PGN) and lipoteichoic acids that bind TLR2.
7.3.1.1 Gut Microbiota, Gut Barrier Integrity, and Chronic Low-Grade Inflammation
The gut barrier consists of an epithelial monolayer and its intercellular junctional system, mucus, and mucosal immune system.
Intestinal epithelial barrier integrity is an important factor in innate immunity function. It is essentially controlled by a complex system of junction proteins (tight junctions, zonula adherens, gap junctions, and desmosomes). The apical junction system is represented by tight junctions (TJs) and the zonula adherens. Together, the TJs and zonula adherens form the “apical junction complex” (AJC). In addition to providing a barrier to noxious molecules, TJs can also operate as pores for the permeation of ions, solutes, and water, as appropriate. The interplay among the tetra-span proteins (occludin, claudin, and tricellulin) [41] and specific claudin isoforms (claudin-2, 7, 12, and 15) determines selective barrier permeability. Single-span transmembrane proteins, on the other hand, are mostly junctional adhesion molecules. Belonging to the immunoglobulin superfamily, junctional adhesion molecules participate in the regulation and maintenance of the TJ barrier in an active way. These proteins are associated with peripheral membrane (plaque) proteins such as zonula occludens (ZO) proteins 1, 2, and 3 (Fig. 7.3) [42].
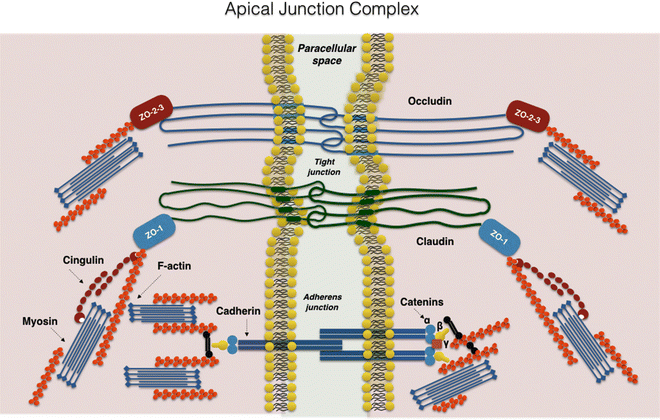
Fig. 7.3
The apical junction complex. A simplified diagram of the molecular composition of tight junctions and adherens junctions
On the intracellular side of the plasma membrane, these proteins contribute to anchoring the junctional protein complex to the actin component of the cytoskeleton, making it sensitive to all factors acting both on the latter and on mitochondrial oxidative phosphorylation, such as FANS and propionic pump inhibitors [43].
The AJC complex is regularly remodeled by interactions with external stimuli: food, pathogenic bacteria, and microbiota components.
The mucosal integrity is guaranteed by the continuity of the epithelial cells and the junctional structure. Additionally, Paneth cells are a specialized type of secretory epithelial cells that inhabit the mucosal surface of the small intestine and produce high quantities of defensins, and other antimicrobial and antibiotic peptides that are very important for correct and ambient intestinal homeostasis [44]. With Paneth cell secretion, the goblet cells are also important for the production of the mucus consisting of a set of glycosylated molecules and enzymes. The mucus comprises two stratified layers: the external layer creates an ideal culture medium for microbial colonization, the inner layer, a denser layer, contains relatively few bacteria and exerts a protective function for the host [44].
The augmented paracellular permeability of the small intestine allowed the passage (leakage) of small quantities of microbes, and endotoxins in the circulation, such as LPS [45]) and peptidoglycan [46], with a secondary activation of the inflammatory response in target tissues such as liver, adipose tissue, coronary vessels, and articulations by triggering PRRs. It follows the onset of various diseases that together develop the framework of the MS.
The increased intestinal permeability promotes LPS absorption, which leads to a state of low-grade metabolic endotoxemia characterized by serum LPS concentration 10–50 times lower than the typical values for septicemia. This induces a state of chronic low-grade inflammation (CLGI) known as leaky gut syndrome (LGS).
Several studies suggest that the host–microbiota relationship might lead to the modulation of the gut barrier function [15, 47, 48].
The gut microbiota may modulate the intestinal barrier integrity via several mechanisms. The intestinal microbiome, by its metabolism of vegetable polysaccharides and short-chain fatty acids (SCFAs) production can regulate the GLP-2 production, improving mucosal barrier function. In effect, administration in mice of prebiotics increases the production of GLP2 in the jejunum and colon via both autocrine and paracrine regulatory loops, reducing metabolic endotoxemia, hepatic inflammation, and oxidative stress markers [47].
The role of microbiota in the modulation of gut permeability could also be mediated by the action of SCFAs on fatty acid synthase (FAS), an essential enzyme for de novo lipogenesis of the intestinal epithelial cells. FAS helps to maintain the mucus barrier by regulating mucin 2, the dominant mucin in the colon and an important component of the mucus. Indeed, specific modulation of gut microbiota composition with prebiotics, as inulin, improves gut barrier integrity, reduces metabolic endotoxemia, and lowers inflammation [49].
Diabetes, hyperlipidemia, obesity, insulin resistance, hypertension, cardiovascular disease, and arteriosclerosis are metabolic illnesses configuring the metabolic syndrome characterized by systemic CLGI [50]. Several findings correlate the altered intestinal permeability with the state of metabolic endotoxemia and consequent systemic low-grade inflammation [51] linked with a gradual deterioration of the organs involved in the inflammatory process.
The HF diet caused an increment of intestinal permeability by microbiota as demonstrated by the use of antibiotic treatment that abolished diet-induced gut permeability [13, 15]. Indeed, it is now well accepted that HF diet-induced metabolic diseases are associated with low-grade inflammation occurring, among other tissues, in adipose depots. This inflammatory process can cause an increase in inflammatory cytokine production and it is also characterized by increased macrophage infiltration [52].
Several research works demonstrate that an HF diet and consequent microbiota composition changes can contribute to the disruption of the TJ proteins (zonula occludens-1 and occludin) involved in the gut barrier function [51]. Moreover, some studies have demonstrated that intestinal luminal exposure to oleic acid can cause intestinal epithelial damage [53]. These effects induced by an HF diet could be mediated through the microbiota and TLR-2.
This condition plays an important regulatory role in tight junctions associated with intestinal epithelial barrier integrity. TLR-2 deficiency predisposes to alterations of TJ-modulated barrier function leading to the perpetuation of mucosal inflammation [40, 54]. Caricilli et al. [55] demonstrated that TLR2 knockout mice present an increase in LPS absorption secondary to decreased expression of TJ protein zonula occludens (ZO)-1 in the ileum with consequent disorder of gut barrier functions. This could be determined by a decrease in Bifidobacterium spp. The authors observed TLR2 knockout mice with a decrease in Bifidobacterium spp., supporting the lower expression of zonula occludens 1 (ZO-1). After gut microbiota transplantation from TLR2 knockout to Bacillus-monoassociated wild-type mice, Caricilli et al. [55] observed a reduction in the expression of ZO-1 in the ileum of the recipients, suggesting that the expression of this protein might be regulated by the particular microbiota present in the gastrointestinal tract of TLR2 knockout mice.
An HF diet promotes intestinal absorption of bacterial endotoxins by different mechanisms demonstrated in several findings. LPS can cross the intestinal barrier by transcellular transport through intestinal epithelial cells and/or intestinal-epithelial microfold cells (M-cells). These specialized epithelial cells are present in the intestinal epithelium; they are also permeable to bacteria and macromolecules, and able to express gut antigens via the underlying lymphoid tissue.
Amar et al. [56] demonstrated that an HF diet induces a “low-grade bacteremia” by an augmented bacterial translocation from the intestinal mucosal barrier. The authors showed that both the adipose tissue and the blood from diabetic mice fed with an HF diet contain living bacteria. They originated from the intestine and were linked to low-grade inflammation. Furthermore, intestinal mucosa from HF-fed mice exhibits properties that are different from the mucosa of normal chow-fed mice and which facilitate bacterial adherence. Using a labeled E. coli bacterial strain with a fluorophore and an ampicillin reporter gene, Amar et al. [56] believed that an HF diet led to an increase in the E. coli co-localization with dendritic cells (DCs) in the intestinal lamina propria. The bacteria, probably remaining inside the DCs, then rapidly disseminated into mesenteric adipose tissue (MAT) and corresponding mesenteric lymph nodes (MLN). However, the presence of various bacteria in MAT and corresponding MLN represents a physiological mechanism that is exacerbated during HF diet-induced metabolic diseases. It is noteworthy that an HF diet increases the accumulation of Gram-negative bacteria that produce LPS, which are highly inflammatogenic molecules, by triggering TLR4 in different tissue.
Toll-like receptors are expressed on the apical surface of enterocytes, where they recognize the MAMPs and active binding and internalizing of these endotoxins [57]. Indeed, the enterocytes can internalize Gram-negative bacteria through TLR4, which mediates phagocytosis and translocation of Gram-negative bacteria in vivo [58]. Furthermore, LPS may be internalized by enterocytes via a myeloid differentiation protein-2 (MD-2)-dependent mechanism [58]. In fact, MD-2 has been recognized as a key molecule for LPS signaling [59].
An HF diet seems to favor absorption of LPS because these have a great affinity for chylomicrons. There is a possibility that LPS could be associated with chylomicrons within the enterocyte and then secreted from cell-associated pools in a chylomicron-dependent manner. This condition could induce continuous postprandial low-grade endotoxemia finally resulting in low-grade inflammation [60] Indeed, TLR4 binding LPS could pass through the intestinal barrier assembled in chylomicrons [61]. Additionally, LPS is internalized in enterocytes through a TLR4 and is transported to the Golgi compartment and finally secreted from the basolateral surface into chylomicrons (Fig. 7.4) [62].
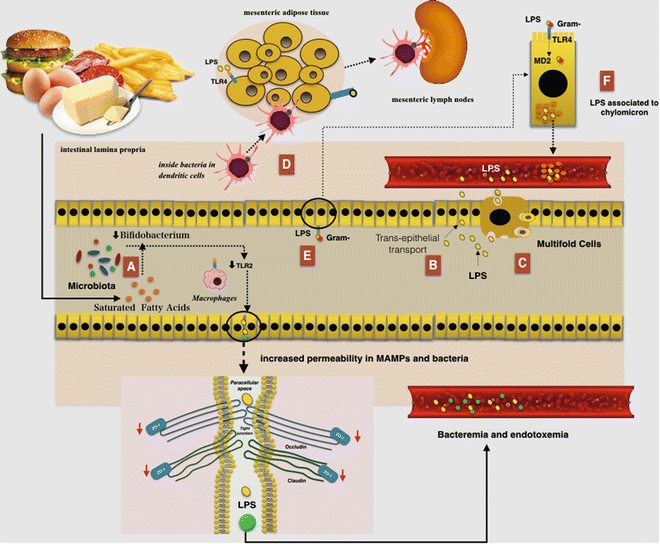
Fig. 7.4
The relationship between a high-fat (HF) diet and augmented gut permeability to bacteria and microbial-associated molecular patterns (MAMPs): (a) A reduced number of Bifidobacterium could induce TLR2 expression and then a reduction in the expression of zonula occludens 1 (ZO-1) would result in alterations of tight junction (TJ)-modulated barrier function, leading to the perpetuation of mucosal inflammation. (b) Lipopolysaccharides (LPS) can cross the intestinal barrier via transcellular transport through the intestinal epithelial cells. (c) LPS can also cross the intestinal barrier via transcellular transport through microfold cells. (d) An HF diet led to an increase in the E. coli co-localization with dendritic cells (DCs) in the intestinal lamina propria. The bacteria, probably remaining inside the DCs, are then rapidly disseminated into mesenteric adipose tissue and corresponding mesenteric lymph nodes. (e) The TLR is expressed on the apical surface of enterocytes and mainly recognize the MAMPs, where it is capable of binding and internalizing purified endotoxin and bacteria. Furthermore LPS may be internalized by enterocytes through a myeloid differentiation protein-2 (MD-2)-dependent mechanism. (f) LPS have a great affinity for chylomicrons and thus could be associated with them within the enterocytes and then secreted from cell-associated pools in a chylomicron-dependent manner
7.3.2 The Extraintestinal Causes of Increased Gut Permeability
7.3.2.1 Stress Impairs Gut Permeability
Numerous environmental factors are able to impair the gut permeability. Many of those act both through stress and activation of the hypothalamic–pituitary–adrenal (HPA) axis. Indeed, both cognitive and noncognitive stressors activate HPA axis inducing many important effects on gut. Noncognitive stressors, similar to pathogens, activate stress by IL-1β, IL-6, and TNFα secretion throughout inflammatory cells induction. These cytokines directly stimulate hypothalamus corticotropin-releasing factor (CRF) secretion, activating the HPA axis. In this way LGCI induces stress and increases LGS levels. Also, prolonged cognitive (psychological) stressors, such as chronic work stress, are linked to higher levels of cytokines IL-6 and TNFα [63]. Stressed mice demonstrate enhanced splenic macrophage microbicidal activity, a sign of increased innate immunity activity [64]. Exposure to cognitive stressors also increases cytokine production via LPS-stimulated splenic macrophages through the effects of stress on gut permeability to MAMPs [64].
The role of microbiota in this phenomena is demonstrated by the absence of this effect in germ-free mice. Furthermore, antibiotic treatment attenuating the stressor-induced splenic macrophage reactivity and preventing the increased circulating levels of bacterial peptidoglycan (PGN), suggest that translocation of microbiota-derived PGN might promotes innate immune system activation.
Several indications have demonstrated increased sympathetic nervous system (SNS) activity as being responsible for enhanced innate immune activity [65].
Exposure to stress results in alteration of the MGBA inducing important alteration in gut physiology: (1) alteration in gastrointestinal motility; (2) increase in visceral perception; (3) changes in gastrointestinal secretion; (4) increase in intestinal permeability; (5) negative effects on regenerative capacity of gastrointestinal mucosa and mucosal blood flow; (6) alteration of interactions between the immune system of the host and microbiota, inducing dysbiosis [66].
The brain interacts with the gut through multiple pathways, including the vagal and splanchnic pathways, HPA axis, and other connections. All of these comprised the gut–brain axis (GBA) and for its important interplay with microbiota: the microbiota–gut–brain axis (MGBA). Indeed, as stress modifies the bacterial flora of the gut, inducing an immunological alteration of the GALT, at the same time, microbiota may have an important effect on the GBAm acting on modulation of the motility.
The involvement of the MGBA in the stress is particularly induced by corticotrophin releasing factor (CRF). The CRF family peptides are expressed both in the CNS and within the gut. Here, CRF plays an important role in many biological actions: inflammation modulation, visceral hypersensitivity (increasing pain perception), augmentation of gut permeability, and motility modulation.
An important pathogenetic role in induction of the gut alteration in stress is played by the mast cells. These cells, which have receptors for CRF on their surface, translate many signals of stress to the intestine [67]. Several studies demonstrate that CRF induces mast cell activation, causing the release of many pro-inflammatory mediators, including de novo synthesized mediators such as prostaglandins and leukotrienes, or preformed granule-housed mediators, including histamine, serine proteases, tryptase, chymase, and finally cytokines such as TNFα [68]. By the release of these mediators, mast cells induce increased intestinal permeability [68, 69]. In an animal model, Overman et al. [70] demonstrated that the gut barrier damage induced by mast cell activation via CRF is mediated by TNFα and proteases. Furthermore, CRF-mast cell signaling pathways and increased intestinal permeability require critical input from the enteric nervous system. In an animal model CRF could inhibit NLRP6 inflammasome signaling, which, with NLRP3, is important in the maintenance of healthy microbiota through the secretion of IL-18 [71]. The consequent alteration of microbiota composition stimulates CCL5 expression in intestinal epithelial cells, resulting in augmented permeability and adsorption of bacterial products and microorganism passage through the paracellular space. Administration of probiotics prevents stress-induced mucosal damage, demonstrating the pathogenetic role of dysbiosis caused by inhibition of NLRP6 via CRF (Fig. 7.5).
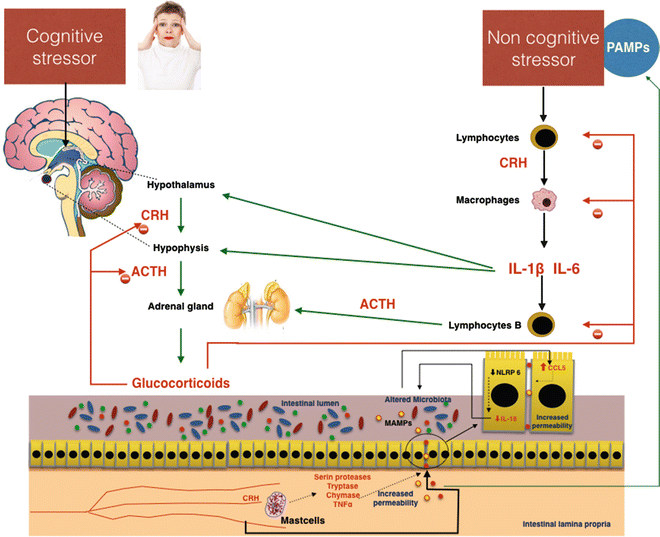
Fig. 7.5
Possible effects of cognitive and noncognitive stressors on gut permeability. Important effects altering intestinal permeability are mediated by corticotropin-releasing hormone (CRH), which is directly stimulated by cognitive stress and indirectly by noncognitive stressors through the mediation of proinflammatory cytokines IL-6 and IL1β. In the gut, CRH activates secretion of proteases and TNFα in mast cells. These substances damage the intestinal barrier, inducing increased permeability. The passage of pathogen-associated molecular patterns (PAMPs) and bacteria in the blood circulation increases the stress, favoring the development of systemic low-grade inflammation. ACTH adrenocorticotropic hormone
Stress induces a very important change in microbiota composition, in neurotransmitter and proinflammatory cytokine levels with direct and indirect effects on the same microbiota. Bailey et al. [65] demonstrated that stressor exposure has an impact on the stability of the microbiota and leads to bacterial translocation across the gut barrier. Mice subjected to chronic stress, which increases circulating cytokines and primes the innate immune system for enhanced reactivity, showed significant changes in the community structure of the microbiota with a decrease in Bacteroides and an increase in Clostridium. The stressed mice also showed increased circulating levels of IL-6 and MCP-1, which correlated significantly with stressor-induced changes to three bacterial genera (Coprococcus, Pseudobutyrivibrio, and Dorea). Mice treated with antibiotics did not show increased IL-6 and MCP-1 levels; dysbiosis is the primary cause of increased circulating cytokine levels.
The evidence suggests a paradigm in which stressor exposure alters homeostatic interactions among the intestinal microbiota, the mucosal immune system and balance in the MGBA. This leads to gut barrier damage and to translocation of MAMPs and pathogenic and/or commensal microbes from the lumen of the intestines to the interior of the host, where they trigger systemic inflammatory responses, inducing LGCI and anxiety-like behavior.
7.3.2.2 Iatrogenic Damage of Intestinal Permeability
Several therapeutic treatments may impair gut permeability. These include radiotherapy and many pharmacological therapies using antibiotic, chemotherapeutic, immunosuppressive drugs and nonsteroidal anti-inflammatory drugs (NSAIDs). These play a fundamental pathogenetic role in damage of the gut barrier. Indeed, these are over-the-counter drugs that are also widely used by heart patients. According to AIFA (Italian agency for drugs) statistics, the defined daily dose (DDD) of prescribed NSAIDs per 1,000 inhabitants per day in 2014 in Italy was 22.8.1 This indicates that 2.28% of the Italian population (about 1,360,000 people) receive a daily prescription of NSAIDs from physicians. Recent studies demonstrate that damage of the mucosal barrier induced by NSAIDs is exacerbated by the use of proton pump inhibitors (PPIs).
It is generally accepted that the use of NSAIDs is frequently prone to side effects on the gastrointestinal system. Early pathogenetic events include a “topical” phase in addition to the inhibition of cyclooxygenase, followed by a multistage pathogenetic event in which intestinal permeability is impaired until awareness of gastric ulcers and intestinal erosions [72, 73]. NSAIDs can determine significant damage to the intestinal mucosa causing both increased intestinal permeability and erosive lesions through pathogenetic mechanisms acting at topical and systemic levels. Somasundaram et al. [73] demonstrated that within 1 h after administration of indomethacin rat intestine is damaged at the brush border of intestinal cells and their mitochondria are vacuolated. NSAIDs act at two levels: (1) systemic, inhibiting cyclooxygenase and reducing the modulatory effects of PGs on intestinal homeostasis; (2) local, independent of the effect on cyclooxygenase and induced by the acidity of NSAIDs and its ability to uncouple oxidative phosphorylation and inhibit electron transportation into the mitochondria. NSAID administration results in a slowing down of mitochondrial respiration via uncoupling oxidative phosphorylation, as shown by the reversal of effect depending on the concentration. Indeed, NSAIDs stimulate mitochondrial respiration (uncoupled oxidative phosphorylation) at concentrations between 30 and 1,500 μM, depending on the particular NSAID studied, and inhibit respiration at higher concentrations [73].
Side effects of NSAIDs on the gut barrier are increased when they are associated with a low dose of aspirin, as in heart patients with chronic articular pain. In these cases, the intestinal damage score is higher than using NSAIDs alone. Surprisingly, the intestinal damage score rises when NSAIDs are associated with PPI and much higher if administered with PPIs and low-dose aspirin [74].
7.3.2.3 Gluten and Gut Permeability
During the past few years there has been a remarkable increase in the popularity of a gluten-free diet. Many people believe that it improves their health and leads to the disappearance of symptoms related to different diseases that share an inflammatory state. This attracted the interest of researchers to a disorder related to the ingestion of gluten or gluten-containing cereals, namely nonceliac gluten sensitivity (NCGS). This disorder was originally described in the 1980s [75] and is characterized by intestinal and extra-intestinal symptoms related to the ingestion of gluten-containing food in subjects who are not affected by either celiac disease (CD) or wheat allergy (WA). Thus, NCGS is a de facto gluten-related disorder. Indeed, to enable a consensus on new nomenclature and the classification of gluten-related disorders, a panel of experts first met in London in February 2011. The meeting resulted in a series of definitions and developed a diagnostic algorithm that has recently been published [76].
The most important pathogenetic mechanism induced by gluten is related to its capacity to increase gut barrier permeability. Although a recent study conducted by Sapone et al. [77] demonstrated in NCGS patients the absence of increased gut permeability, gluten is one of the more powerful factors able to trigger zonulin release and innate immune response.
Zonulin system (ZS) is an immunologically related, endogenous modulator of epithelial TJs [78]. Tripathi et al. [79] characterized zonulin as pre-haptoglobin2 (pre-HP2); to date, only regarded as an inactive precursor for HP2, one of two genetic variants of human haptoglobins (HP1 and HP2). The physiological role of this pathway is not completely clear. It appears to be involved in several functions, including TJ regulation responsible for the movement of fluid, macromolecules, and leukocytes between the bloodstream and the intestinal lumen, and vice versa [80]. Another potential physiological role is in innate immunity protecting against microorganism colonization of the proximal intestine [81]. The structure of zonulin is similar to several growth factors, which like zonulin, affect TJ integrity [82]. Indeed, zonulin activates epidermal growth factor receptor (EGFR), causing TJ disassembly, and then augments gut permeability [79]. Zonulin as pre-HP2, in its intact configuration, could activate EGFR through direct binding and/or through protease activated receptor 2 (PAR2), a transmembrane receptor that modulates inflammatory responses and acts as a sensor for proteolytic enzymes generated during infection. Intestinal tryptase IV induces structural changes in the molecule, altering its ability to bind to EGFR, but enabling its function as a hemoglobin scavenger. Meanwhile, it becomes an inflammatory marker [83]. Barone et al. [84] demonstrated that gliadin acts like EGF on actin cytoskeleton, with effects very similar to those induced by zonulin. Therefore, gliadin induces zonulin release by intestinal cells and whole intestinal tissues through CXCR3 binding (Fig. 7.6) [27, 28, 81].
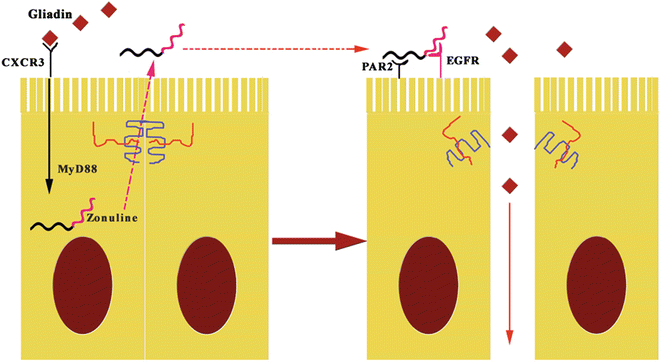
Fig. 7.6
The pathogenetic effects of gliadin on gut permeability are mediated by zonulin. The fragments 11–130 and 151–170 of gluten binding to CXCR3 activates zonulin secretion via MyD88, enabling paracellular translocation of gliadin through the effects of zonulin on the TJ, increasing paracellular permeability. EGFR epidermal growth factor receptor
Gluten is a complex macromolecule consisting of a globular protein, gliadin, and a fibrous protein, glutenin.
There are at least 50 toxic epitopes in gluten peptides with cytotoxic (peptide fragment 31–43), immunomodulatory (33-mer gliadin fragment – 57–89 peptide and IL8-releasing peptide – fragment 261–277), and gut-permeating activities (CXCR3-binding zonulin-releasing peptide fragments 11–130 and 151–170) [85].
The permeabilizing effects of gliadin peptides in vivo was confirmed in patients with active celiac disease (CD) and non-CD control patients, when analyzing their intestinal tissue after gliadin challenge [86]. The Fasano group studies suggests that gluten induces changes in gut permeability and activation of innate immunity response comes before that of adaptive immunity [87].
According Fasano, gliadin ingestion throughout IL8-releasing peptide induces [88] IL-8 release and recruitment of neutrophils in the intestinal lamina propria in CD patients. Together, CXCR3-binding zonulin-releasing fragments activates zonulin secretion through MyD88 pathway enabling paracellular translocation of gliadin. Consequently, 33-mer gliadin and other immunomodulatory peptides of gliadin interact with macrophages of gut submucosa [89]. This interaction via MyD88 induces Th1-type cytokine secretion, resulting in mononuclear cell infiltration of intestinal submucosa. The presence of IFNγ and TNFα augments epithelial and endothelial gut permeability [90].
In genetically predisposed subjects (DQ2+ and DQ8+) gliadin antigen presentation by dendritic cells to Th cells in intestinal lamina propria induces the loss of oral tolerance and switch to Th1/Th17 response. The migration of gliadin-loaded DCs in mesenteric lymph nodes leads to migration of CD4 − CD8 − γδ, and CD4 − CD8 + αβ T cells to the gut, where they cause inflammation in patients with CD.
7.3.3 CLGI in Cardiovascular Disease
The role of CLGI in the pathogenesis of arteriosclerosis and other CVDs is now clear and accepted. Currently, the main challenge in atherosclerosis research is the identification of disease-specific PAMPs or DAMPs able to give innate immune responses in the arterial wall, and finally to promote plaque development [91]. In 1999, Wiedermann demonstrated how subclinical endotoxemia constitutes a strong risk factor for the development of carotid atherosclerosis, particularly among smokers [92]. The same study revealed that plasma from individuals with prevalent atherosclerosis of the carotid arteries could activate endothelium, promoting leukocyte transmigration; this suggested the role of systemic pro-inflammatory mediators. Furthermore, plasma collected from patients without atherosclerosis at the time of enrollment in the study was tested for its capacity to induce endothelial cell activation and transmigration of leukocytes. Increased plasma-induced endothelial cell activation was associated with an increased risk for the development of atherosclerotic lesions such as revealed by subsequent follow-up data on new lesion formation after 5 years. These data indicated that plasma-mediated endothelium activation by interaction with leukocytes promotes the development of atherosclerotic lesions. Plasma collected from patients without atherosclerosis at the time of enrollment in the study resulted in being able to induce endothelial cell activation and transmigration of leukocytes in those subjects who had subsequently developed atherosclerotic lesions [93].
It has long been recognized that in sepsis caused by Gram-negative bacteria, plasma endotoxins are elevated and able to evoke inflammatory cells, such as monocytes and macrophages, and in endothelial and smooth muscle cells numerous proinflammatory responses, including up-regulation of adhesion molecule expression, increased production of cytokines, reactive oxygen species (ROS), and reactive nitrogen species, prostaglandins, and tissue factor, loss of monolayer integrity and barrier function, and apoptosis. However, endotoxin levels observed during sepsis are far greater than the low-level endotoxemia that has been associated with atherosclerosis. The Bruneck study demonstrated that subjects with levels of 50 pg/mL or greater were recognized as being at an increased risk for the development of atherosclerosis. This level of endotoxin can induce inflammatory responses in human monocytes and macrophages and intact human blood vessels exhibit profound responsiveness to endotoxins (cytokine release, superoxide production, and monocyte adhesion). Low oxidative stress modulates endothelial gene expression, which induces atherogenic factors, forming early atherosclerotic plaque [94].
Serum lipoproteins, especially high-density lipoprotein (HDL), play a major role in the clearance of circulating endo- toxin [95]. HDL binds LPS and transports them to the liver hepatocytes and hepatic macrophages provide LPS clearance and its final biliary excretion [95]. Low-density lipoprotein (LDL) is generally believed to be much less effective than HDL at removing endotoxin from the blood. Moreover, in a study conducted in human volunteers, infusion of HDL was associated with reduced LPS-mediated release of TNF-α, IL-6, and IL-8 [96].
Innate immunity throughout its receptor system plays an important role. Endotoxins are able, via TLR activation, to promote different types of proinflammatory responses strictly connected to the pathogenesis of atherosclerosis. In phagocytic cells, endotoxins potently induce ROS production by stimulating NADPH oxidase activity. This enzyme is present in other vascular cells, too, such as endothelial cells, smooth cells, fibroblasts, and may participate in the induction of cytokine expression, smooth muscle cell growth and apoptosis. It can play an important role in the pathogenesis of hypertension and atherosclerosis [97]. The oxidant process induced by ROS induces a vicious circle via lipid peroxidation with formation of lipoperoxides that behave like ROS, amplifying oxidative damage. The oxLDL acts as a TLR4 agonist on macrophages. Indeed, the formation of the plaque is initiated through the uptake of LDL from blood by endothelial cells, and the oxidation of LDL by reactive oxygen species within the vessel wall. Subsequently, the oxLDL is phagocytized by macrophages through scavenger receptors (such as CD36 and macrophage scavenger receptor class A (SR-A) leading to macrophage transformation into lipid-laden activated foam cells. Foam cells produce proinflammatory cytokines and matrix metalloproteinases and express costimulatory molecules for T-cell activation (such as CD40). The recruitment of lymphocytes at the inflammatory site of the vessel induces the production of IgM-specific antibodies for oxLDL during atherogenesis. The formation of immune complexes could have protective effects on foam cell formation. Indeed, these antibodies may be able to cross the endothelial monolayer to reach the atherosclerotic plaques. There, they bind to antigens on the surface of oxLDL, form immune complexes, and block the uptake of oxLDL by macrophages.
The stimulation of TLR4 on vascular smooth cells induces an increased LDL and extracellular matrix deposition in the plaque. IFN-α enhances TLR4 signaling and thereby the consequent production of TNF-α, IL-12, and matrix metalloproteinase 9 (MMP9) with amplification of the inflammatory process [98].
Induction of proinflammatory cytokines and chemokines by PRR stimulation is associated with plaque pathogenesis. LPS binding protein, CD14, toll-like receptor-4 (TLR-4), and MD-2 at the cell membrane initiate the inflammatory cascade that throughout NF-kB recruits proinflammatory cytokines and chemokines at the inflammatory site [99–101]. Of the earliest cytokine discoveries among those implicated in the pathogenesis of arteriosclerosis, there is macrophage migration inhibitory factor (MIF), which has since been shown to be expressed by monocytes/macrophages, T cells, B cells, and by endocrine and epithelial cells [102]. MIF exerts both autocrine and paracrine effects on TLR-4 expression. In addition to the induction of MIF, endotoxins can contribute either directly or indirectly to increased release of other cytokines including IFNγ, IL-1, IL-6, IL-8, TNFα, and granulocyte-macrophage colony- stimulating factor (GMCSF), along with platelet-activating factor (PGF) [103]. It is very important that endotoxin also contributes to the increased expression of the TNF receptor [104].
Of the chemokines induced by triggering PRRs via endotoxins, MCP-1 and IL-8 appear to play critical roles in atherosclerosis. The first is highly expressed in human atherosclerotic plaques and plays a crucial role in monocyte recruitment into subendothelial lesions of vessels [105]. MCP-1 induction is sensitive to very low levels of endotoxin (1 ng/mL) [106, 107]. IL-8 is known to be chemotactic for neutrophils. IL8 activates NADPH oxidase in these cells and increases production of ROS, initiating the oxidative process [108]. This cytokine also plays a role as a chemotactic factor, recruiting at the site of the lesion monocytes and T-lymphocytes that are prevalent in the fibrous cap of atherosclerotic lesions, where they may be involved in the pathogenesis of acute coronary syndromes. Low levels of endotoxin induce IL8 release and confirm the role of low-grade endotoxemia in the pathogenesis of CLGI in CVD.
The release of chemokines attracts inflammatory cells to inflammatory sites of the vessel wall where monocytes, neutrophils, and T lymphocytes arrive. The “rolling” of leukocytes on the endothelial surface is mediated by selectins (P and E selectins) [109] (that are not normally expressed on endothelial cells, but their expression can be transcriptionally induced by cytokines, bacterial endotoxins, and other mediators [110]). Next, “firm adhesion” of leukocytes is permitted by the binding of β 2 -integrins expressed on leukocytes to cellular adhesion molecules (CAMs), such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule (VCAM-1), which are expressed on the apical surface of the endothelium [109]. After adhesion, leukocytes cross the endothelium via interactions between PECAM-1 molecules, which are expressed by both cell types [111]. The process of transmigration of the inflammatory cells is modulated by endotoxins at multiple steps. They activate β 2 -integrins, upregulate E-selectin and CAMs, in addition to increasing phosphorylation of platelet endothelial cell adhesion molecule-1 (PECAM-1), the release platelet adhesion factor (PAF), and the expression of its receptor on endothelial cells [109, 112].
A central role in plaque formation is played by macrophages in which there is excessive accumulation of lipids, resulting in foam cell formation. TLR4 stimulation by LPS and/or FFA has been shown to contribute to early-stage intimal foam cell accumulation. Intimal smooth muscle cells surround and penetrate early lesions, where TLR4 signaling, enhanced by hypercholesterolemia, promotes lesion progression by stimulation of acyl-coenzyme A, cholesterol acyltransferase-1 mRNA expression, cytoplasmic cholesterol ester accumulation, and MCP-1 mRNA and protein expression in a TLR4-dependent manner [113]. The modulation of endotoxin-induced cellular activation could be one mechanism for the antiatherogenic effects of high HDL levels. Indeed, a recent study in humans has shown that HDL reduces plasma levels of TNF-α and expression of CD11b on monocytes [114].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


